|
|
CHAPITRE XVI
DIFFRACTION DES RAYONS X
Détermination des paramètres moléculaires
(Pour les applications, voir un cours de radiocristallographie - Rayons X)
|
Les chapitres précédents traitent de l’interaction entre la lumière et les molécules que ce soit en l’absence ou en présence de champs électrique ou magnétique. On le sait la lumière est un phénomène caractérisé par son aspect ondulatoire comme par exemple dans le cas de la diffraction. À un faisceau de rayons X correspond un comportement ondulatoire. Question :
Objectif :
|
La cristallographie a pour but de déterminer la symétrie cristalline à partir de la position q des réflexions. Elle nous apprend à relier chaque raie observée à une famille de plans réticulaires caractérisés par les indices de MILLER h, k, l. Si le réseau est formé d’une seule catégorie d’atomes, l’analyse aux rayons X est terminée lorsqu’on a déterminé la maille élémentaire. Si le réseau est moléculaire on introduit dans la maille un motif composé de l’ensemble des atomes de la molécule avec leurs positions respectives par rapport à un atome de base que l’on peut choisir arbitrairement.
Chacun des atomes du motif et l’ensemble de ses homologues dans le cristal sont aux nœuds d’un réseau simple qui n’est autre que le réseau du cristal. Le cristal peut donc être considéré comme formé par la superposition d’un certain nombre de réseaux simples se déduisant les uns des autres par des translations. Si le rayon incident n’a pas la direction d’une réflexion sélective pour le réseau simple, les ondes diffusées par les atomes aux nœuds de chacun des réseaux se détruisent complètement par interférences. Par conséquent, l’onde diffusée par le cristal entier sera aussi nulle. Il ne peut donc pas exister pour le cristal avec motif d’autres directions de réflexions que celles du réseau simple. Le motif introduit dans le réseau n’a pas d’influence sur la position des faisceaux diffractés. Nous allons voir que son seul effet est d’en modifier les intensités.
Illustrons par un exemple simple cette notion très importante. À chaque atome du motif correspond un réseau simple, donc, une famille de plans réticulaires de même écartement d. Entre deux plans réticulaires A, A1 successifs correspondant à un atome donné, s’intercalent les plans où se trouvent les homologues des autres atomes de la maille A', A", A"'. Soit un rayon incident sous l’angle tel que 2 d sin q = n l.
Chaque groupe de plans Ai', Ai", Ai''' donne des ondes en phase. L’amplitude de l’onde diffractée par tout le cristal sera égale à N fois la somme de l’amplitude des ondes diffusées par le groupe de plans A, A', A" contenue entre A et A1, si N est le nombre de plans A dans le cristal. Soient a, a', les amplitudes diffusées par chacun des plans du groupe. Elles peuvent être différentes si la nature des atomes ou leur densité dans le plan varie d’un plan à l’autre. Soient j';j"; j"'. Les différences de phases entre les ondes diffusées par les plans A et A' et A" et A"'. La construction géométrique classique de FRESNEL permet de trouver l’onde résultante. L’intensité de cette onde résultante (carré de son amplitude) va évidemment dépendre de l’amplitude des ondes élémentaires et de leur différence de phase. Cette différence de phase dépend uniquement d’un facteur géométrique : la position dans l’espace des différents atomes composant la molécule.
En résumé l’intensité est proportionnelle, si on néglige l’absorption dans le cristal :
Le produit de ces deux coefficients est appelé facteur de structure. De la mesure des intensités, on pourra déduire le facteur de structure expérimental. Le facteur de structure expérimental pourra être comparé à un facteur de structure calculé à partir d’un modèle hypothétique.
16.2 Coefficient de diffusion atomique
Les rayons X sont diffusés presque entièrement par les électrons et l’intensité de la radiation diffusée par un atome particulier doit dépendre de la distribution de tous les électrons dans l’atome. Pour les petits angles de diffraction l’amplitude du faisceau diffusé est égale à la somme des amplitudes des faisceaux qui seraient diffusés par chaque électron individuel. Dans ces conditions, l’amplitude totale est proportionnelle au nombre d’électrons. Le facteur de diffusion atomique pourra donc être pris égal au nombre d’électrons. Pour des angles de diffraction plus grands les différents rayons diffusés vont interférer, la diffusion sera réduite et le facteur deviendra plus petit que le nombre d’électrons.
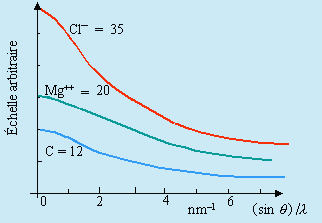
Les facteurs de diffusion atomique peuvent être calculés à partir des fonctions d’onde électroniques. Les résultats sont trouvés dans les tables (Fig. 16.1).
|
|
Figure 16.1. Coefficients de diffusion atomique.
L’intensité d’un faisceau diffracté par un plan d’atomes est proportionnelle au coefficient de diffusion atomique.
Le coefficient géométrique dont il a été question dans l’introduction peut se calculer. On sait que lors de la réflexion correspondant à une petite série de plans h, k, l, les rayons sur deux plans consécutifs correspondant à l’atome A sont en phase c’est-à-dire que leur différence de phase est 2 p. Si l’atome A est lié à un atome B (molécule A B) il existe une série de plans h, k, l, contenant ces atomes déduits de la première série par la translation A.B. Si on considère un plan h, k, l contenant deux atomes A et le plan h, k, l, parallèle voisin contenant l’atome B, on montre par un calcul géométrique que la différence de phase entre ces deux plans est :
![]()
Dans cette équation x, y et z sont les coordonnées de B par rapport aux axes cristallographiques. Le facteur de diffusion atomique de A étant fA et fB, l’onde résultante diffractée par la molécule A B sera la somme géométrique de l’onde diffractée par A et de l’onde diffractée par B. La construction de FRESNEL donne l’onde résultante puisqu’on connaît la différence de phase entre ces deux ondes.
![]()
L’intensité est proportionnelle à ce facteur.
Généralisons cette étude à une molécule possédant N atomes i dont les coordonnées sont xi, yi, zi et le facteur de diffusion fi. Le plan h, k, l, portant les atomes i diffracte les rayons en introduisant la différence de phase.
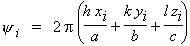
par rapport au plan h, k, l portant les atomes i = i.
La construction de FRESNEL appliquée aux N ondes de phase ji donne :
![]()
L’intensité est proportionnelle à ce facteur. Le terme Fhkl est le facteur de structure. Il fait intervenir le pouvoir diffusant de chaque atome de la molécule ainsi que sa position dans l’espace par rapport à un atome de base choisi comme origine.
16.4 Méthode de détermination des paramètres moléculaires
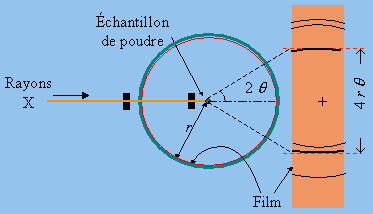
Le principe de la méthode peut être exposé de façon simple. Le diagramme de diffraction nécessaire aux études chimiques est obtenu le plus souvent par la méthode des poudres (DEBYE-SCHERRER - Fig. 16.2). Ce diagramme permet d’indexer les raies, c’est-à-dire de déterminer à quel plan h, k, l appartient chaque raie. D’autre part, on peut mesurer l’intensité de chaque raie.
|
|
Figure 16.2. Description de la méthode des poudres.
Mesure de l’intensité des raies
La plupart des analyses ont été faites en estimant visuellement les densités sur les films photographiques au moyen d’un film Standard. Ce film standard est fait, de préférence avec le même cristal en l’exposant pendant des durées différentes. L’effet provoqué sur le film par les intensités I et 2I par exemple est le même que celui provoqué par la même intensité pendant les temps t et 2t. L’œil peut facilement comparer des noircissements de films identiques, donc finalement, les intensités. On peut de cette façon dresser un tableau des intensités de raies à l’aide d’une échelle arbitraire, c’est-à-dire obtenir des nombres proportionnels aux intensités. L’erreur est de l’ordre de 20 %. Le compteur GEIGER maintenant utilisé donne une précision de 3 % mais il faut déplacer le compteur de façon à mesurer dans chaque raie séparément ce qui prend beaucoup de temps. Cette méthode est maintenant généralisée et permet des enregistrements automatiques suivies par traitement ordinateur, parfois en temps réel, des résultats expérimentaux.
Correction des intensités
Chaque nombre de l’échelle d’intensité obtenue précédemment est proportionnel au facteur de structure. Les intensités dépendent toutefois d’autres facteurs moins importants sans doute mais non négligeables. Il se produit une absorption et une polarisation des rayons X dans l’échantillon. Cette absorption dépend de l’angle de diffraction et du coefficient d’absorption A de la matière. L’absorption peut se traduire par un coefficient de la forme :
|
A g(q) où g(q) = |
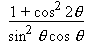 |
la variation du facteur d’absorption étant faible on peut le négliger.
L’intensité est d’autre part proportionnelle au nombre de plans de la famille considérée. Cette proportionnalité se traduit par le facteur p; facteur de multiplicité. C’est le nombre de plans dont les indices h, k, l sont différents mais pour lesquels d (h, k, l) est le même.
Finalement, l’intensité peut se mettre sous la forme :
|
Ihkl = k p A g(q) |
|
k est un coefficient de proportionnalité constant pour toutes les raies d’un même cristal et il ne dépend plus que de l’intensité du faisceau X incident.
Si on veut effectuer une comparaison des intensités X diffractées il faut faire intervenir ces facteurs. Compte tenu de ces facteurs, on obtient une nouvelle série de nombres qui sont uniquement proportionnels au carré du facteur de structure. La racine carrée de ces nombres donne le facteur de structure observé
![]()
Il faut ensuite postuler un modèle moléculaire et calculer ce facteur de structure calculé,
![]()
en fonction de la position xi, yi et zi des divers atomes. Si le facteur calculé et le facteur observé ne concordent pas, il faut déplacer les atomes dans le modèle de façon à réaliser l’accord. Cette méthode d’approche pourrait être très longue (puisque chaque atome peut prendre une infinité de positions) si la symétrie de la molécule n’était pas déjà connue à l’aide d’autres méthodes: stéréochimie classique, moment dipolaire, spectroscopie infrarouge. Cette méthode est cependant limitée aux cristaux les plus simples.
16.5 Analyse des densités électroniques par la méthode de FOURIER
Prévue par BRAGG et mise au point par PATTERSON, la méthode précédente est applicable aux structures les plus simples et nécessite la connaissance de la position des atomes.
Une méthode utilisant directement les résultats expérimentaux c’est-à-dire les intensités corrigées, a été proposée par BRAGG. Elle est actuellement très utilisée pour les grosses molécules.
Un phénomène quelconque, fonction périodique d’une coordonnée peut être représentée par une série de FOURIER
F(x) = A0 + A1 cos 2p [x/a + a1] + A2 cos 2p [x/a + a2] + . . .
![]()
A est l’amplitude de chaque onde, a la phase et a la distance de répétition du phénomène. L’équation précédente en une dimension peut être étendue aux trois dimensions.
On peut précisément considérer que la densité électronique se répète dans le volume du cristal, avec les distances de répétition a, b, c, dimensions de la cellule élémentaire cette densité électronique peut donc se mettre sous la forme d’une triple série de FOURIER.
Les amplitudes des ondelettes Ahkl sont évidemment liées au facteur de structure puisque l’intensité de la diffusion est liée à la densité électronique et à la phase. On montre finalement que l’on a :
![]()
Cette équation permet en principe de résoudre une structure quelconque. Il suffit de connaître le volume V de la cellule élémentaire et le facteur de structure observé F.
Les séries de FOURIER peuvent alors être évaluées pour chaque point x y z, et donneront une carte de la densité électronique dans la matière. Aux positions réelles des atomes, la densité électronique sera maximum alors qu’elle sera zéro entre les atomes. En principes, il faudrait calculer une infinité de termes pour chaque série de FOURIER. Pratiquement, ces termes tendant rapidement vers zéro, il suffit de calculer les premiers.
On obtient ainsi des cartes de la densité électronique déterminant la position des atomes. Cette densité dépend également du nombre d’électrons de l’atome et permet d’identifier ce dernier. Cette méthode ignore complètement l’atome d’hydrogène, car sa contribution à la diffusion est trop faible puisqu’il ne possède qu’un seul électron.
La diffraction X est une méthode très puissante d’analyse des structures. La taille de la molécule ne semble pas être une limite. La structure de la vitamine B 12, soit près de 200 atomes, a été résolue avec succès. La structure de la pénicilline a été un triomphe de la cristallographie puisqu’elle fût déterminée avant que les organiciens ne connussent la formule développée. Certains chercheurs sont actuellement engagés dans des analyses de structure de molécules comportant plus de mille atomes.
Tout comme la lumière, la diffraction d’un faisceau de rayons X permet d’obtenir des informations précieuses sur les structures, particulièrement des structures cristallines (voir un cours de cristallographie). Il faut se rappeler que la longueur d’onde de ces faisceaux est de l’ordre de grandeur des distances inter nucléaires facilitant ainsi les interactions.
|
Pour en savoir un peu plus
Lien intéressant :
Toujours à l'index sous les expressions "Fourier analysis" et "Bragg's law" du site http://hyperphysics.phy-astr.gsu.edu/hbase/hframe.html (visité le 2019-02-27).
Dernière mise à jour : 2021-07-07.
_______________________________