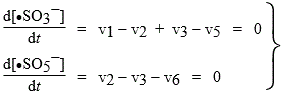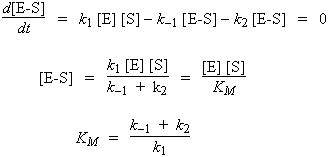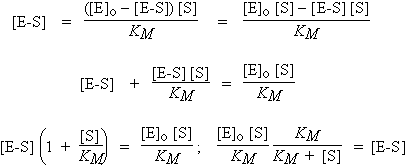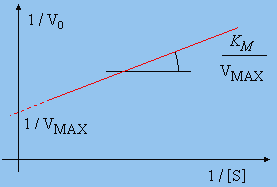|
|
CHAPITRE 8
LES RÉACTIONS CATALYSÉES
Préambule/Objectifs Les réactions chimiques entre diverses entités sont donc gouvernées par des lois où l’énergie joue un rôle prépondérant. Questions :
|
Objectifs spécifiques
- Comprendre les mécanismes de catalyse (d’inhibition) de mécanismes réactionnels.
- Comprendre l’influence du catalyseur (de l’inhibiteur) sur la barrière de potentiel.
- Maîtriser un exemple de mécanismes impliqués dans le fonctionnement d’une horloge chimique.
- Comprendre la spécificité et les variantes possibles de la catalyse enzymatique.
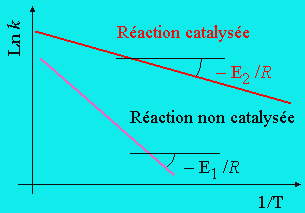
On a déjà vu que les vitesses de réactions obéissent à des lois bien précises. La thermodynamique y joue un rôle important. Deux facteurs interviennent plus spécifiquement : le facteur pré-exponentiel, A, et l’énergie d’activation, Ea. Ces deux facteurs convenablement réunis traduisent la constante de vitesse.
( k = A e-Ea/RT )
Cette constante de vitesse n’est valable que pour un système donné. À partir d’un ensemble de réactifs, via un chemin réactionnel approprié, le système évolue, réagit vers la formation des produits. Cette constante de vitesse dépend du chemin suivi puisque Ea mesure la hauteur de la barrière de potentiel. Contrairement à la thermodynamique qui ne tient pas compte du chemin suivi (principe de l’état initial et de l’état final), la cinétique chimique est largement dépendante du chemin suivi. En modifiant ce chemin, on modifiera du même coup la barrière de potentiel. Si l’on augmente Ea, la barrière s’élève et la constante de vitesse décroît. Inversement si l’on diminue Ea, la barrière se fait moins haute et la constante de vitesse s’accroît.
Une manière relativement simple pour modifier cette barrière est l’usage de catalyseurs. En principe, un catalyseur, ajouté à un système réactionnel, aide au déroulement de la réaction sans être lui-même consommé. En fait, il participe à certaines étapes élémentaires, mais est régénéré. Il permet aux réactifs de trouver un chemin réactionnel moins difficile en abaissant la hauteur de la barrière de potentiel. Le chemin réactionnel suivi dans un système non-catalysé est le chemin directe : il devient détourné dans un système catalysé. Pour les fins de ce chapitre, on ne parlera que de la catalyse en phase homogène, se réservant pour plus tard les phénomènes mettant en jeu plusieurs phases.
|
|
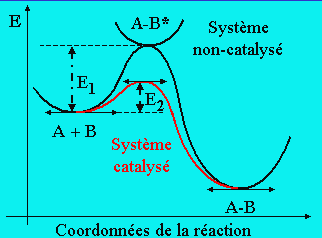
Figure 8.1. Coordonnées réactionnelles d’un système catalysé ou non catalysé.
Un cas simple de catalyse homogène a déjà été signalé dans le chapitre 3 à propos de la dégénérescence d’ordre. L’hydratation de l’isobutène est facilitée par la présence d’ions H+. Plus généralement, soit le système
A + B ® A-B s’effectuant avec une énergie d’activation E1
La même réaction en présence de catalyseur, C, s’écrit :
A + B + C ® A-B + C s’effectuant avec une énergie d’activation E2
Pour le moment, on ne sait rien du mode d’action de C. La seule chose que l’on observe est une augmentation de la vitesse de formation du produit A¾B. En admettant que l’énergie d’activation E2 est inférieure à E1, on obtient une explication de l’augmentation de la vitesse : figures 8.1 et 8.2. En général, le facteur pré-exponentiel est beaucoup moins perturbé que l’énergie d’activation, de telle sorte que l’on peut ignorer cet effet tout au moins en première approximation.
Exemple d’une réaction catalysée :
Soit la décomposition thermique de l’ozone catalysée par le chlore :
| [1] | Cl2 + O3 | ® ClO• + ClO2• | k1, initiation |
| [2] | ClO2• + O3 | ® ClO3• + O2 | k2, propagation |
| [3] | ClO3• + O3 | ® ClO2• + 2 O2 | k3, propagation |
| [4] | 2 ClO3• | ® Cl2 + 3 O2 | k4, rupture de chaîne |
|
|
Figure 8.2. Énergie d’activation d’un système catalysé.
ClO2· et ClO3· sont les porteurs de chaîne auxquels on peut appliquer le principe de quasi-stationnarité. ClO· se décompose en chlore et oxygène atomiques, lesquels ne participent pas vraiment à la chaîne et finissent par redonner le chlore et l’oxygène moléculaire. La vitesse de disparition de l’ozone est telle que :
| = v1 + v2 + v3 = k1 [Cl2] [O3] + [O3] (k2 [ClO2•] + k3 [ClO3•]) |
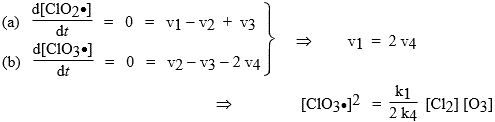
Si la chaîne cinétique est importante, la réaction [4] est quantitativement peu importante devant la réaction [2] : v4 << v2 et v2 = v3 :
| Þ | [Cl2]1/2 [O3]3/2 + k1 [Cl2] [O3] |
Comme on a un mécanisme en chaîne, v1 << v2 + v3 en ce qui regarde la disparition de l’ozone :
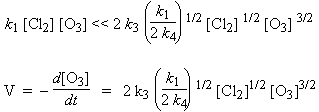
Puisque la pression partielle de
chlore est constante, V = a [O3]3/2. La constante de
proportionnalité comprend la concentration du catalyseur. Si la longueur de
chaîne, ![]() , est le rapport entre le nombre de fois que se répète l’étape
de propagation de chaîne sur le nombre de fois que se produit l’étape d’amorçage,
, est le rapport entre le nombre de fois que se répète l’étape
de propagation de chaîne sur le nombre de fois que se produit l’étape d’amorçage,
![]() , est :
, est :
|
En simplifiant, |
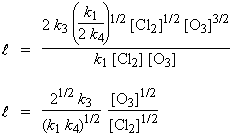 |
La longueur de chaîne,
![]() , est
d’autant plus longue que la concentration du catalyseur est
faible. En d’autres termes, plus il y a de catalyseur et plus la concentration de porteurs
de chaîne croît, augmentant du même coup la probabilité de la réaction de rupture de
chaîne. En conséquence, un catalyseur n’est pas nécessairement d’autant plus efficace
que sa concentration est grande. En fait, vitesse de réaction et longueur de chaîne ne
sont pas des paramètres interchangeables.
, est
d’autant plus longue que la concentration du catalyseur est
faible. En d’autres termes, plus il y a de catalyseur et plus la concentration de porteurs
de chaîne croît, augmentant du même coup la probabilité de la réaction de rupture de
chaîne. En conséquence, un catalyseur n’est pas nécessairement d’autant plus efficace
que sa concentration est grande. En fait, vitesse de réaction et longueur de chaîne ne
sont pas des paramètres interchangeables.
Un catalyseur augmente donc la vitesse de réaction. Existe-t-il des catalyseurs "négatifs" ? En d’autres termes, existe-t-il des composés qui augmentent la barrière de potentiel ? Cette question, bien que théoriquement acceptable, est triviale. Un système réactionnel prendra toujours le chemin le plus facile. Le fait d’ajouter un inhibiteur ne modifie pas la barrière de potentiel de la réaction considérée. Par contre, sa présence permet d’ouvrir d’autres chemins réactionnels plus faciles. Le système réactionnel est alors orienté vers une autre "sortie" de telle sorte qu’il se produit une diminution de la vitesse de la réaction originellement considérée.
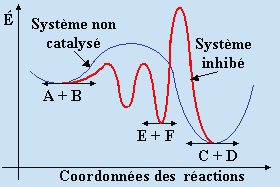
On a déjà vu un tel système dans le cas de la photoisomérisation du cis-2-butène catalysée par l’hydrogène sulfureux (Chapitre 5). La présence de 1,3-butadiène intercepte les porteurs de chaîne que sont les radicaux •SH. Il y a véritablement synthèse de dérivés soufrés de nature vinylique. Les porteurs de chaîne sont donc orientés vers une autre voie réactionnelle : figure 8.3.
|
|
Figure 8.3. Représentation d’un système inhibé.
Dans certains cas, la disparition de l’inhibiteur permet au système réactionnel de retrouver son allure originelle. Le système réactionnel est véritablement en présence d’un inhibiteur de réaction.
Exemple d’une réaction inhibée :
Soit la réaction de synthèse du chlorure d’hydrogène à partir de l’hydrogène et du chlore en présence d’oxygène :
| [1] | Cl2 + M | ® 2 Cl• + M | amorçage |
| [2] | Cl• + H2 | ® HCl + H• | propagation |
| [3] | H• + Cl2 | ® HCl + Cl• | propagation |
| [4] | H• + O2 + M | ® HO2• + M | rupture de chaîne |
| [5] | Cl• + O2 + M | ® ClO2• + M | rupture de chaîne |
| [6] | 2 HO2• | ® H2O2 + O2 | rupture de chaîne |
| [7] | H2O2 | ® H2O + 1/2 O2 |
Les deux dernières réactions sont sans effet sur le mécanisme puisque la rupture de chaîne est le fait des réactions [4] et [5] qui sont relativement lentes. En appliquant le principe de quasi-stationnarité aux espèces atomiques •H et •Cl, on obtient :
d[•Cl]/dt = 2 v1 - v2 + v3 - v5 et d[H•]/dt = v2 - v3 - v4
En résolvant simultanément ces deux équations, on obtient les valeurs de [•H] et de [•Cl] :
| et |
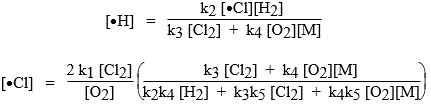 |
La vitesse de réaction est :
V = d[HCl] / dt = v2 + v3
| V = k2[•Cl][H2] + k3[•H][Cl2] = [•Cl] | 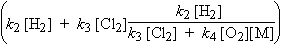 |
En substituant [•Cl] par sa valeur, en réarrangeant, et en supposant que les réactions incluses dans la chaîne sont beaucoup plus importantes que les réactions de terminaison (qui sont des réactions thermoléculaires, donc à vitesse lente sous pression normale), l’expression précédente se simplifie :
![]()
Comme on peut le voir, l’oxygène réagit avec les porteurs de chaîne. La vitesse de formation de HCl diminue en présence d’oxygène, comme le montre l’équation de vitesse. C’est un inhibiteur en ce sens qu’il oriente certains porteurs de chaîne vers d’autres réactions. Dans le cas de la synthèse de HBr, le radical BrO2• n’existant pas, l’oxygène n’a que très peu d’effet.
Autre
exemple de réaction catalysée
Soit
l’isomérisation du cis-2-butène en
trans-2-butène. Cette
réaction requiert la réalisation de la rotation autour de la double liaison.
Cette réaction a une barrière de potentiel élevée : DH
> 400 kJ/mole.
En présence d’atomes d’iode, ceux-ci s’additionne de manière réversible
sur la double liaison. La liaison
double fait place à une liaison simple qui se prête bien à la rotation,
abaissant d’autant la barrière de potentiel : : DH
< 20 kJ/mole.
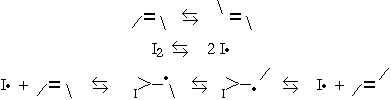
4. Réaction induite
Soit les réactions d’oxydation des ions Mn++ et As+++ par un chromate HCrO4-. L’oxydation des ions Mn++ par un chromate est une réaction lente alors que celle de l’acide arsénieux, H3AsO3 (As+++), en acide arsénique, H3AsO4 (As5+), est rapide :
| [1] H3AsO3 + HCrO4- |
|
H3AsO4 + HCrO3- |
| [2] 2 HCrO3- + H3AsO3 + 8 H+ |
|
H3AsO4 + 2 Cr+++ + 5 H2O |
L’oxydation des ions Mn++, en présence
de l’acide arsénieux, est une réaction très rapide. Il semble que l’ion As+++
soit un inducteur pour la réaction d’oxydation des ions Mn++ par l’ion HCrO4-.
Les deux réactions précédentes sont suivies de :
[3] HCrO3-
+ Mn++ + 5 H+ ® Mn+++ + Cr+++
+ 3 H2O
soit au total [1] + [3] :
[4] HCrO4- + H3AsO3 + Mn++
+ 5 H+ ® H3AsO4 + Mn+++ +
Cr+++ + 3 H2O
Autre exemple :
Soit la réaction d’oxydation
d’un hydrocarbure par l’eau oxygénée. Le mélange RH + H2O2
n’est
pas très réactif. On considère en effet que l’eau oxygénée est un oxydant très
léger. Si l’on ajoute du sulfate ferreux on observe une augmentation importante de la
vitesse d’oxydation. Le couple FeSO4 + H2O2 constitue un
mélange très oxydant, bien que pris séparément, les deux additifs soient plutôt
neutres : il y a synergie importante.
[5] H2O2
+ Fe++ ®
•OH + Fe+++
+ OH-
[6] •OH
+ Fe++ ® Fe+++ + OH-
C’est la réaction classique de
l’oxydation du fer ferreux, Fe++, par l’eau oxygénée. Ces étapes constituent
la période d’induction de l’oxydation des hydrocarbures.
[7] •OH
+ RH ® H2O + R•
[8] R•
+ H2O2 ®
•OH + ROH
De faibles quantités d’ions ferreux provoquent l’oxydation d’un grand nombre de molécules d’hydrocarbure à travers un mécanisme en chaîne linéaire. Les ions ferreux ne constituent pas à proprement parler un catalyseur puisqu’ils sont consommés : ils deviennent des ions ferriques, Fe+++. Cependant, ils provoquent la formation de porteurs de chaîne convenables pour l’oxydation de l’hydrocarbure. On parlera de préférence d’un accélérateur de réaction ou d’induction de réaction.
Autre exemple : Oxydation de l’acide oxalique par le chlore induite par les ions Fe++ :
| [9] | Fe++ + Cl2 | ® •Cl + Fe+++ + Cl- | amorçage, | |
| [10] | •Cl + C2O4H- | ® Cl- + •C2O4- + H+ | propagation, | |
| [11] | •C2O4- + Cl2 | ® •Cl + Cl- + 2 CO2 | propagation, | |
| [12] | •Cl + •Cl | ® Cl2 | rupture, | |
| [13] | •Cl + •C2O4- | ® Cl- + 2 CO2 | rupture. |
Globalement (en faisant la somme
des étapes incluses dans la chaîne) :
[14] C2O4H-
+ Cl2 ® 2 Cl- + 2 CO2 + H+
Certains composés produisent eux-mêmes leur propre "catalyseur" de réaction. Il existe ainsi de nombreux produits chimiques qui se détruisent, s’auto transforment, en d’autres produits : on dit aussi qu’il y a auto catalyse. Pour les garder, on doit leur ajouter un stabilisateur que l’on peut considérer comme un inhibiteur réactionnel. Soit par exemple le cas des sulfites. Laissés en présence d’air, les sulfites ont la fâcheuse tendance à s’oxyder dans un mécanisme en chaîne :
| [1] | Cu++ + SO3-- | ® Cu+ + •SO3- | amorçage |
| [2] | O2 + •SO3- | ® •SO5- | propagation |
| [3] | •SO5- + HSO3- | ® HSO5- + •SO3- | propagation |
| [4] | HSO5- + SO3-- | ® HSO4- + SO4-- | réaction rapide |
| [5] | Cu+ + •SO3- | ® Cu++ + SO3-- | rupture |
| [6] | X + •SO5- | ® non- porteurs de chaîne | rupture |
La réaction [5] est l’opposée de la réaction [1]. L’équilibre thermodynamique engendre donc le porteur de chaîne •SO3-. On ajoute un stabilisateur, qui n’est rien d’autre qu’un inhibiteur. Dans ce cas, on utilise de l’alcool, X.
La vitesse de formation des sulfates est telle que :
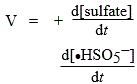 |
= 2 v4 = 2 v3 = 2 k3 [SO5-] [HSO3-] |
| = v4 - v3 |
Appliquons ce principe de quasi-stationnarité aux porteurs de chaîne :
|
|
Þ v1 - v5 - v6 = 0 |
Þ k1 [Cu++] [SO3--] - k5 [Cu+] [•SO3-] - k6 [•SO5-] [X] = 0
| [•SO3-] = |
|
et
k2 [•SO3-] [O2] - k3 [•SO5-] [HSO3-] - k6 [•SO5-] [X] = 0
En remplaçant dans cette dernière expression [•SO3-] par la valeur tout juste obtenue :
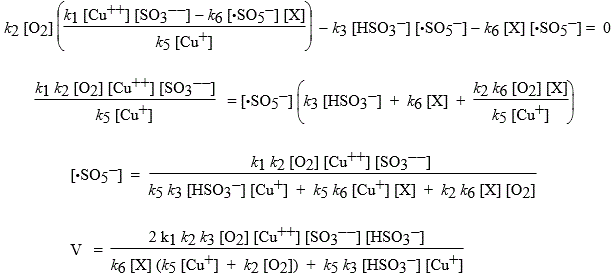
1- Si [X] = 0 ou si l’inhibiteur est peu actif,
![]()
2- Si au contraire, X est très actif :
![]()
L’oxydation est alors très lente.
6. Systèmes oscillants - l'horloge chimique
Quelques systèmes réactionnels présentent des caractéristiques assez particulières. Ainsi, avec des combinaisons relativement bien choisies de réactifs, on a pu mettre en évidence la possibilité de créer des systèmes oscillants, encore appelés horloges chimiques. Soit par exemple une solution acide d’ions bromates. C’est un milieu suffisamment oxydant pour oxyder l’acide malonique en gaz carbonique et en acide formique, avec formation concurrente d’acide bromomalonique. Il a été étudié pour la première fois par BELOUSOV en 1958.
3 BrO3- + 5 CH2(COOH)2 + 3 H+ ® 3 BrCH(COOH)2 + 2 HCOOH + 4 CO2 + 5 H2O
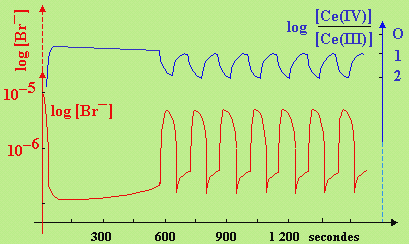
La réaction est très lente, mais elle est catalysée par des sels de cérium. La figure 8.4 montre le comportement de cette solution et plus particulièrement les variations des concentrations des ions Br- et du rapport Ce4+/Ce3+. Après une période d’induction dont la durée dépend des conditions expérimentales, on voit que le système semble évoluer entre deux positions extrêmes : il y a oscillation des concentrations des ions Br- et des ions cérium, avec la régularité quasi-exemplaire d’une horloge. Cette horloge n’est cependant pas éternelle : elle s’arrête lorsque l’un des réactifs est épuisé. Lorsque le système est en présence d’une quantité importante d’ions bromures, la cinétique réactionnelle peut être décrite par le mécanisme suivant :
| [A] | BrO3- + Br- + 2 H+ | ® HBrO2 + HOBr |
| [B] | HBrO2 + Br- + H+ | ® 2 HOBr |
| [C] | 3 HOBr + 3 Br- + 3 H+ | ® 3 Br2 + 3 H2O |
| [D] | 3 Br2 + 3 CH2(COOH)2 | ® 3 BrCH(COOH)2 + 3 Br- + 3 H+ |
| soit globalement : | ||
| [X] | BrO3- + 2 Br- + 3 CH2(COOH)2 + 3 H+ | ® 3 BrCH(COOH)2 + 3 H2O |
|
|
Figure 8.4. Tracés potentiomètriques de log[Br-]
et du log [Ce(IV)]/[Ce(III)] dans une solution homogène constituée d’acide malonique, de
bromure et de bromate de potassium.
en présence d’ions Ce3+ et en milieu H2SO4 [J. Am. Chem. Soc.,
94, 8650 (1972)].
La cinétique est relativement simple puisque la première réaction d’oxydoréduction constitue l’étape limitante :
|
V = - |
|
= kA [BrO3-] [Br-] [H+]2 |
La concentration de HBrO2 peut être considérée constante : ce produit n’apparaît ni ne disparaît. On peut donc écrire (principe de quasi-stationnarité) :
VB = V ou encore :
kA [BrO3-] [Br-] [H+]2 =
kB [HBrO2] [Br-] [H+]
et
[HBrO2] =
(kA/kB)
[BrO3-] [H+]
En présence d’ions Ce(III) et en l’absence d’ions bromures et d’ions Ce(IV), le mécanisme devient le suivant. Bien que les deux premières étapes soient réversibles, seule la réaction [E] impose sa vitesse à l’ensemble.
|
VY = - |
|
= kE [BrO3-] [HBrO2] [H+] |
Les deux réactions [E] et [F] sont évidemment de type auto catalytique et la concentration de HBrO2 croît. La somme de ces deux réactions est la suivante :
| [E] | BrO3- + HBrO2 + H+ | 2 BrO2• + H2O | |
| [E] | 2 BrO2• + 2 Ce3++ 2 H+ | 2 HBrO2 + 2 Ce4+ | |
| (BrO3- + 2 Ce3+ + 3 H+ | HBrO2 + 2 Ce4+ + H2O) | ||
| [G] | 2 HBrO2 | ® | BrO3- + HOBr + H+ |
| [H] | HOBr + CH2(COOH)2 | ® | BrCH(COOH)2 + H2O |
soit globalement :
| [Y] | BrO3- + CH2(COOH)2 + 5 H+ + 4 Ce3+ | ® | BrCH(COOH)2 + 3 H2O + 4 Ce4+ |
| [I] | BrO3- + HBrO2 + 2 Ce3+ + 3 H+ | 2 HBrO2 + 2 Ce4+ + H2O |
Comme la réaction [G] détruit le HBrO2, on atteint alors un état d’équilibre où la concentration de ce produit peut être 100 000 fois plus élevée qu’en absence d’ions Ce(III).
[HBrO2]y = (kE/2 kG) [BrO3-] [H+]
On peut donc voir que la concentration de HBrO2 dépend de la concentration de l’ion Br-. Lorsque cette dernière est grande, la réaction [X] est importante et la valeur de [HBrO2] devient petite. À ce moment, la vitesse de la réaction globale [Y] devient négligeable. Le point critique apparaît lorsque les vitesses des réactions [B] et [E] sont égales.
kB [Br-] = kE [BrO3-]
Pour que le système oscille il faut que la concentration en ion bromure soit reconstituée. Les ions Ce(IV) créés dans la réaction [F] réagissent avec les acides malonique et bromomalonique:
[J] 6 Ce4+ + CH2(COOH)2 + 2 H2O ® 6 Ce3+ + HCOOH + 2 CO2 + 6 H+
[K] 4 Ce4+ + BrCH(COOH)2 + 2 H2O ® Br- + 4 Ce3+ + HCOOH + 2 CO2 + 5 H+
Ces deux réactions deviennent éventuellement rapides. Au fur et à mesure que la concentration en ion Br- augmente, la réaction [B] redevient rapidement importante et, la réaction [I] étant incapable de répondre suffisamment vite, la concentration de HBrO2 décroît rapidement. Le cycle des réactions [A] à [D] redevient important et le cycle recommence.
Produits chimiques :
500 ml d’une solution 1 M de H2SO4;
14,30 g d’acide malonique, HOOC-CH2-COOH;
5,22 g de bromate de potassium, KBrO3;
0,548 g de nitrate double d’ammonium et de cérium (4+), Ce(NH4)2(NO3)6;
1 à 2 ml d’indicateur coloré ferroine.
Mélanger ensemble et attendre quelques minutes pour la période d’induction.
D’autres systèmes oscillants ont été observés. Par exemple, la réaction de BRAY-LIEBHAFSKY implique l’oxydation catalytique du peroxyde d’hydrogène par un iodate :
| 2 IO3- + 5 H2 O2 + 2 H+ | ® I2 + 5 O2 + 6 H2O | |
| I2 + 5 H2O2 | ® 2 IO3- + 4 H2 O + 2 H+ | |
| Globalement : | 5 H2 O2 | ® 5/2 O2 + 5 H2O |
7. La catalyse enzymatique (Cinétique de MICHAELIS-MENTEN)
On appelle ainsi toutes les réactions "biochimiques" spécifiquement catalysées par les enzymes. Rappelons qu’une enzyme est un catalyseur très spécifique puisque son action est beaucoup plus limitée que celle des catalyseurs. Soit le mécanisme :
![]()
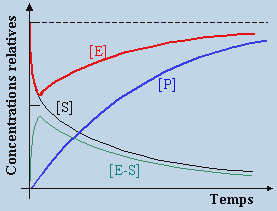
Dans ce système réactionnel, E, S, et P sont respectivement l’enzyme, le substrat et les produits et E-S un complexe enzyme - substrat. En mélangeant initialement une enzyme E à un substrat S, l’équilibre 1 étant très rapide, une fraction importante de l’enzyme s’ajoute sur le substrat, l’importance étant conditionnée par la constante thermodynamique de l’équilibre 1. La décomposition du complexe enzyme-substrat démarre et donne lieu à la formation du produit P en régénérant l’enzyme. La réaction 2 est elle-même partie d’un équilibre thermodynamique. Cependant, en début de réaction, l’absence du produit P et l’abondance du complexe E-S limite considérablement l’importance de la réaction –2. Ces observations sont résumées dans la figure 8.5.
|
|
Figure 8.5. Réaction
enzymatique : variations des concentrations des réactifs
et des produits en fonction du temps de réaction.
En supposant donc que la quantité du produit P est nulle au départ de la réaction et en appliquant le principe de quasi-stationnarité à la concentration du complexe enzyme-substrat :
| Þ |
|
| Þ | |
|
où |
KM est ce qu’on appelle la constante de MICHAELIS.
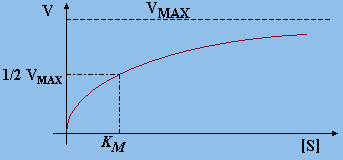
La vitesse initiale de la réaction est :
![]()
|
|
Figure 8.6. Variation de la vitesse d’une réaction
enzymatique
en fonction de la concentration du substrat [S].
Or, la concentration originelle en enzyme est telle que : [E]o = [E] + [E-S]
La concentration [E]o est connue et non pas [E] ou [E-S]. Donc : [E] = [E]o - [E-S]
et
| [A] |
Puisque Vo = k2 [E-S]
| [B] |
|
La vitesse maximum est obtenue lorsque [S] >> KM :
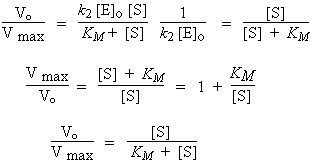 |
|
| ou encore [C] | |
|
Puisque |
Le rapport Vo / Vmax = 1/2 lorsque KM = [S]
|
[D] |
|
|
|
Figure 8.7. Graphe de LINEWEAVER-BURK.
En divisant de part et d’autre l’équation [C] par Vmax , on obtient la relation de LINEWEAVER-BURK :
![]()
Cette relation est aussi connue, sous forme modifiée (après avoir multiplié de part et d’autre par la concentration [S] la relation de LINEWEAVER-BURK), sous le nom de l’équation de DIXON :
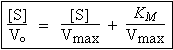
Une autre façon de déterminer les constantes est de multiplier de part et d’autre la relation [D] par la quantité Vmax. On obtient la relation d'EADIE :
Vo Vmax KM + Vo Vmax [S] = Vmax2 [S]
Vo Vmax [S] = Vmax2 [S] - Vo Vmax KM
Vo [S] = Vmax [S] - Vo KM
Vo = Vmax - KM Vo / [S]
Le graphe de Vo en fonction du rapport Vo/[S] donne une droite de pente égale à – KM et d’ordonnée à l’origine égale à Vmax . Ce graphe est aussi connu sous le nom de EADIE – HOFSTEE.
8. Effets inhibiteurs sur la catalyse enzymatique
Certains produits de l’activité enzymatique peuvent intervenir à des stages initiaux de la chaîne de production et ainsi donner un feed-back pour permettre la régularisation de l’activité enzymatique. Les inhibiteurs peuvent agir sur l’enzyme de façon réversible ou irréversible. Il est évident que la formation de complexes réversibles est préférable car elle permet à l’organisme de régénérer l’enzyme lorsque les conditions le demandent. Nous allons voir trois types d’inhibition réversible :
On terminera avec un cas trivial de cinétique irréversible.
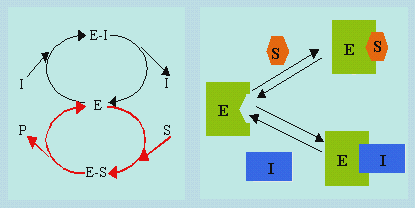
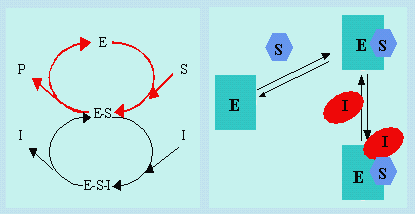
8.1. Inhibiteurs compétitifs
Les inhibiteurs, I, compétitifs réagissent d’une façon réversible avec l’enzyme E pour former des complexes E-I. Le schéma suivant montre le mécanisme impliqué.
|
|
Schéma 8.1. Mécanisme de réaction enzymatique en présence d’un
inhibiteur compétitif.
| E + S | E-S ® E + P | 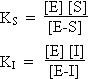 |
|
| E + I | E-I |
KS et KI sont les constantes des équilibres de dissociation des complexes intermédiaires E-S et E-I. Dans le cas de cette inhibition, la concentration totale de l’enzyme s’exprime par :
[E]o = [E] + [E-S] + [E-I]
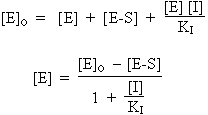 |
|
| ou encore :
|
Dans le cas de l’inhibition compétitive, l’équation devient :
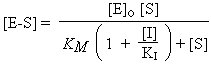
La vitesse de la réaction enzymatique ainsi inhibée s’exprime alors par :
| [E] | 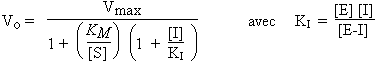 |
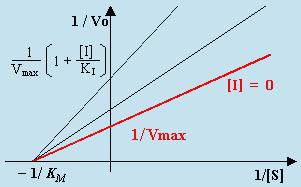
L’équation de LINEWEAVER-BURK devient :
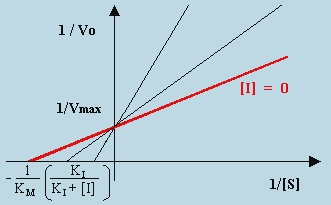
| [F] |
L’intercepte demeure 1/Vmax et la pente est accrue par le facteur (1 + [I]/KI). La valeur de 1/[S] lorsque 1/Vo = 0 est telle que :
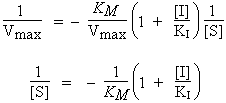
Une caractéristique importante de l’inhibition compétitive réside dans le fait que les droites tracées pour différentes concentrations de l’inhibiteur convergent toutes en un même point sur l’ordonnée à une valeur égale à 1/Vmax. On reconnaît ainsi facilement l’inhibition compétitive (Fig. 8.8).
|
|
Figure 8.8. Graphe de LINEWEAVER-BURK dans le cas de réaction
enzymatique en présence d’un inhibiteur compétitif.
L’inhibiteur entre véritablement en compétition avec le substrat pour réagir avec l’enzyme. Une fois le site occupé par l’inhibiteur, ce site n’est plus, temporairement puisque la réaction est réversible, disponible pour la réaction. La vitesse maximum n’est pas altérée; la vitesse initiale diminue avec une augmentation de la concentration de l’inhibiteur. Par exemple, la réaction de déhydrogénation de l’acide succinique en présence de la déshydrogénase succinique conduit à la formation de l’acide fumarique :
| COOH-CH2-CH2-COOH | trans-COOH-CH=CH-COOH | |
| acide succinique | acide fumarique |
L’acide malonique, COOH-CH2-COOH, est un inhibiteur compétitif car, à cause de sa structure similaire, l’enzyme ne peut différencier l’acide succinique de l’acide malonique. Si on divise l’équation [D] par l’équation [E] :
8.2. Inhibiteurs non-compétitifs
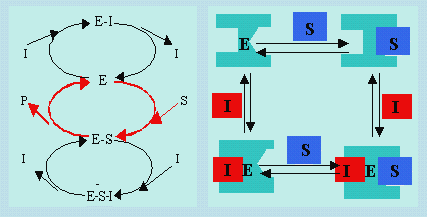
Au mécanisme précédent, s’ajoute une réaction de complexassion réversible entre le complexe enzyme – substrat, E-S, et l’inhibiteur I. Ce dernier intervient donc à la fois sur l’enzyme et sur le complexe E-S. Il faut cependant remarquer que l’inhibiteur n’occupe pas le même site réactionnel que le substrat. Le substrat et l’enzyme se succèdent indifféremment l’un après l’autre pour se lier à l’enzyme.
|
|
Schéma 8.2. Mécanisme de réaction enzymatique en présence d’un
inhibiteur non-compétitif.
Note : si la figure de gauche semble indiquer que le substrat ne réagit qu’avec
l’enzyme, il réagit tout autant avec le complexe enzyme-inhibiteur. La figure de
droite est plus explicite à ce sujet.
| E + S | E-S ® E + P | |
| E + I | E-I | |
| E-S + I | ESI |
Dans le cas de cette inhibition, la concentration totale de l’enzyme s’exprime par :
[G] [E]o = [E] + [E-S] + [E-I] + [ESI]
| où |
|
L’équation [G] se réécrit ainsi :
|
[E]o = [E] + [E-S] |
Il faut remarquer que les réactions de formation des complexes E-S et ESI sont de même nature, les constantes KI et KSI seront vraisemblablement identiques.
| ou encore : | 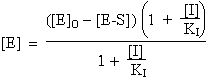 |
Dans le cas de l’inhibition compétitive, l’équation devient :
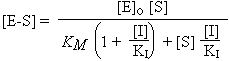
La vitesse initiale de la réaction enzymatique affectée par une inhibition non compétitive s’exprime alors par :
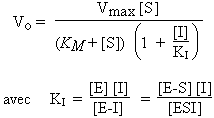
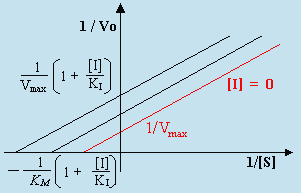
L’équation de LINEWEAVER-BURK devient :
| [H] |
|
Si on trace 1/Vo versus 1/[S], l’intercepte, b, devient :
![]()
et la pente, a, à la droite s’écrit :
![]()
La valeur de 1/[S] lorsque 1/Vo = 0 peut se recalculer :
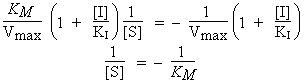
|
|
Une caractéristique importante de l’inhibition non compétitive réside dans le fait que les droites tracées pour différentes concentrations de l’inhibiteur ne convergent pas en un même point de l’ordonnée mais plutôt en un point commun de l’abscisse correspondant à la valeur - 1/KM. On la distingue ainsi de l’inhibition compétitive (Fig. 8.9). Comme dans le cas précédent la vitesse initiale de formation des produits recherchés diminue avec l’augmentation de la concentration en inhibiteur. Par ailleurs, dans le cas présent, la vitesse maximum diminue. Ce type d’inhibition non-compétitive se retrouve pour les groupes SH des cystéines avec les ions de métaux lourds :
| 2 -SH + Hg++ | S-Hg-S + 2 H+ | |
|
-SH + Ag |
-S-Ag + H+ |
8.3. Inhibiteurs incompétitifs
Les inhibiteurs incompétitifs ne se lient pas avec l’enzyme seul. Ils se lient sur le complexe enzyme-substrat pour empêcher la formation du produit. L’effet de l’inhibiteur ne peut être contrecarré par une augmentation de substrat.
|
|
Schéma 8.3. Mécanisme de réaction enzymatique en présence d’un inhibiteur
incompétitif.
| E + S | E-S ® E + P | |
| E-S + I | ESI |
L’analyse du mécanisme réactionnel donne :
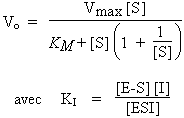
Le graphique de LINEWEAVER-BURK se réécrit de la manière suivante :
| [I] |
|
Si on trace 1/Vo versus 1/[S], l’intercepte, b, devient :
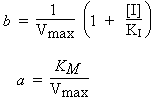 |
|
| et la pente, a, à la droite s’écrit : |
La valeur de 1/[S] lorsque 1/Vo = 0 peut se recalculer :
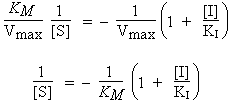
|
|
Figure 8.10. Graphe de LINEWEAVER-BURK dans le cas de réaction
enzymatique en présence d’un inhibiteur incompétitif.
Dans ce cas, la vitesse initiale ainsi que la vitesse maximum diminuent avec une augmentation de la concentration en inhibiteur. L’inhibition incompétitive se retrouve rarement pour les enzymes spécifiques à un substrat. Par contre, les enzymes pouvant réagir avec plusieurs substrats donnent ce type de cinétique en présence d’inhibiteurs.
8.4. Cinétique irréversible
Les équations de Michaelis-Menten ne peuvent être utilisées dans le cas ou l’enzyme et l’inhibiteur forment des complexes irréversibles. L’efficacité des inhibiteurs non réversibles ne dépend évidemment pas de leurs constantes d’équilibres mais de la rapidité à laquelle les inhibiteurs forment des complexes avec l’enzyme.
|
1- Un mécanisme possible de l’hydrogénation catalysée par la vapeur de mercure est le suivant :
[1] Hg + H2 ® Hg + 2 •H, avec sa constante de vitesse k1
[2] •H + C2H4 ® •C2H5, avec sa constante de vitesse k2
[3] •C2H5 + H2 ® C2H6 + •H, avec sa constante de vitesse k3
[4] •H + •H ® H2, avec sa constante de vitesse k4
Déterminez :
Que pensez-vous de la réaction [4] ?
2- L’hydrolyse du sucrose en présence de l’enzyme invertase montre les résultats suivants :
| [S] mol·dm-3 | 0,0292 | 0,0584 | 0,0876 | 0,117 | 0,146 | 0,175 | 0,234 |
| Vitesse (?) | 0,182 | 0,265 | 0,311 | 0,330 | 0,349 | 0,376 | 0,381 |
Tracez le graphe de Lineweaver-Burk; calculez la vitesse maximum ainsi que la constante de Michaelis.
3- L’enzyme fumarase catalyse la réaction suivante :
fumarate + H2O ®
![]() -maléate
-maléate
À 25 °C, avec une concentration [Eo] = 10-6 m, les vitesses de réaction, v, à différentes concentrations de substrat, sont :
| [S] 106 moles | 1,0 | 2,0 | 3,8 | 5,0 | 8,0 | 10 | 20 | 50 |
| v 104 mol·s-1 | 2,6 | 4,3 | 6,3 | 7,2 | 8,7 | 9,3 | 10,8 | 12,0 |
Calculez la constante de Michaelis et la vitesse maximum de la réaction.
4- L'ajout d'une substance à la réaction décrite dans le problème précédent montre qu'elle possède un effet inhibiteur sur la catalyse enzymatique du succinate de sodium. Les données relatives à cette inhibition étant les suivantes, déterminer quel est le type d'inhibition en cause.
|
[Succinate de Na]o |
Vitesse initiale mmol L-1 sec-1 |
|
|
mol L-1 ´ 103 |
[I] = 0,50 mmol L-1 |
[I] = 0,25 mmol L-1 |
|
10,2 |
1,12 |
1,13 |
|
2,0 |
0,87 |
0,92 |
|
1,0 |
0,67 |
0,75 |
|
0,5 |
0,47 |
0,55 |
|
0,33 |
0,36 |
0,43 |
5- Dans la réaction de synthèse du chlorure d’hydrogène inhibée par l’oxygène et en utilisant le principe de quasi-stationnarité, retrouver les expressions des concentrations en atomes d’hydrogène et de chlore ainsi que les coefficients A et B de la vitesse de formation de HCl.
Pour en savoir un peu plus :
Alain Dumon, La naissance des concepts de la cinétique chimique au XIXe siècle, L'atualité chimique, Société chimique de France, N° 436, 41-45 (janvier 2019).
Lewis, E. S., éditeur, Investigation of Rates and Mechanisms of Reactions Part I, 3e édition, Wiley Interscience Publ., J. Wiley & Sons, New York, 1974.
Systèmes oscillants, voir :
numéro spécial, J. Phys. Chem., 93(7), (1989); QD 1 J866.
et pour l’horloge chimique de Landolt, voir :
Gáspár, V. et K. Showalter, J. Am. Chem. Soc., 109, 4869 (1987); QD 1 A512. idem : J. Phys. Chem., 94, 4973 (1990).
Sur le NET :
On ne doit pas se priver de faire des recherches sur le Net tout en maintenant un regard critique sur la qualité des sites observés. Privilégier les sites gouvernementaux, universitaires, les grandes industries, les Fondations, ...
Un site très bien fait avec une approche moderne de la cinétique préparé par M. Dominique Lavabre et Mme Véronique Pimienta : http://pagesperso-orange.fr/cinet.chim/cours/chap6.html (site visité le 2020-11-06).
Dernière mise à jour: 2021-07-02.
x x x x x x x x x x x