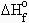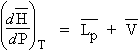| Définitions de systèmes fermé et isolé, S : |
CHAPITRE II
|
Comment peut-on mesurer l’énergie interne d’un système ? Et d’abord, comment définir un système ? Est-ce qu’un système peut seulement échanger de la chaleur avec l’extérieur ? S’il y a variation de volume au cours d’une transformation, cette variation de volume doit commander un certain travail qui s’ajoute algébriquement à l’échange de chaleur. Comment peut-on tenir compte de cet échange de travail ? |
1. Postulat et quelques définitions
Quelques définitions :
-
Variable thermodynamique : ce sont les quantités observables en
thermodynamiques;
- Fonction thermodynamique : fonction qui implique les variables
thermodynamiques;
- Coordonnées thermodynamiques : synonymes de variables thermodynamiques;
- Fonction d’état : fonction thermodynamique qui prend une seule valeur
pour un état donné.
- État d’une substance : liste de toutes ses propriétés physiques et
chimiques.
Quatre paramètres sont nécessaires pour définir thermodynamiquement une substance pure. Toutes les autres propriétés peuvent se déduire (tout au moins en théorie) de ces quatre propriétés. On choisi habituellement la pression P, la température T, le volume V et le nombre de moles n. Mêmes ces quatre paramètres ne sont pas indépendants ; ils sont reliés par une équation appelée équation d’état. Par exemple, pour un gaz parfait le produit pression volume est constant : PV = n RT. Les autres propriétés peuvent être obtenues à l’aide d’opérations appliquées à l’équation d’état.
Postulat :
|
|
Conséquence, l’énergie de l’univers est constante.
| Définitions de systèmes fermé et isolé, S : |
1.1 Système ouvert : échange de matière et d'énergie avec l'extérieur;
1.2 Système fermé : étanche à la matière mais est perméable à l’énergie;
1.3 Système isolé : étanche à la matière et à la chaleur.
2. L’énergie interne d’un système
C’est la quantité d’énergie présente à l’intérieur d’un système : c'est une fonction d'état. Cette quantité est inaccessible. Rien ne permet de la comptabiliser. Pour ce faire, il faudrait mesurer ou avoir accès à toutes les formes d’énergies connues et inconnues. On peut cependant se faire une idée de la façon avec laquelle les molécules peuvent emmagasiner de l’énergie. Les réservoirs d’énergie peuvent être de nature translationnelle (énergie cinétique), rotationnelle (la molécule tourne sur elle-même comme une toupie), vibrationnelle, électronique, magnétique, ...
[2.1] Eint = Si(Etransl + Erot + Evibr + Eélectr + Emagn + ...)
Dans cette équation, i représente chaque molécule comprise dans le système.
3. Variation d’énergie interne d’un système fermé
Si l’énergie interne d’un système isolé n’est pas mesurable, il n’en est pas de même d’un système non isolé. Dans un tel système, on peut avoir accès à la variation d’énergie interne en contrôlant les échanges d’énergie avec l’extérieur. On peut comptabiliser négativement l’énergie qui sort du système et positivement l’énergie qui y entre. Soit par exemple, le système
H2 (g) + 1/2 O2 (g) ® H2O (g)
Cette réaction dégage, libère 240,58 kJ·mol-1 au cours de la formation isométrique d’une mole d’eau gazeuse. Ce dégagement d’énergie se fait au détriment de l’énergie interne du système. Il faut ajouter que le système doit être mieux défini que seulement par une (1) mole d’hydrogène gazeux et une demi-mole d’oxygène gazeux. Il faut aussi préciser si la réaction a lieu à volume ou à pression constante. Si la réaction se fait à volume constant, dans une bombe calorimétrique, il n’y a aucun échange d’énergie sous forme de travail avec l’extérieur. Si au contraire, la réaction a lieu à la pression atmosphérique, donc à pression constante, le volume diminue au cours de la réaction de 33,6 litres à 22,4 litres, soit par un facteur de 1,5. Il y a alors échange de travail avec l’extérieur. Oublions momentanément ce cas pour retenir seulement la réaction à volume constant. L’énergie libérée, mesurée dans un calorimètre par exemple, s’est faite au détriment de l’énergie interne du système. L'énergie interne est une fonction d'état. Sa variation DE est telle que :
DE = - 240,6 kJ·mol-1 de H2O (g) formée.
Si au contraire, la réaction se fait à pression constante, il faut aussi tenir compte des forces de pression et comptabiliser le travail ainsi libéré. La convention de signe est inverse de celle retenue pour l’échange de chaleur.
|
échange de chaleur |
| échange de travail |
Si q = 0, le processus est dit adiabatique. Le travail effectué lors d’une modification de volume (de V' à V") - travail effectué par S sur le milieu- est donné par la relation
|
|
(voir un premier cours de thermodynamique). |
Si le volume V est constant, w = 0 et si le volume n'est pas constant, DV ¹ 0, Þ w ¹ 0 .
Dans le cas d’une réaction s’effectuant sans changement de volume - on dit aussi que la réaction est isochore - la variation d'énergie DE = QV.
Par exemple, la chloration du méthane CH4 se fait sans changement de volume :
CH4 (g) + Cl2 (g) ® HCl (g) + CH3Cl (g); QV = - 107 kJ·mol-1 .
L’énergie interne du système diminue de 107 kJ·mol-1 pour chaque mole de méthane consommée.
4. L’enthalpie
Cependant, la grande majorité des réactions se font à la pression atmosphérique, donc à pression constante - on dit ici que les réactions sont isobares - et
| [2.2] |
|
= q - PV1 + PV2 |
C’était le cas de la réaction H2 (g) + 1/2 O2 (g) ® H2O (g) . On définit alors une nouvelle fonction d'état : l’enthalpie H, telle que H = E + PV . Tout comme l’énergie interne E, H n’est pas susceptible d’être mesuré. Par contre sa variation, DH, peut l’être.
[2.3] DH = DE + D(PV) et comme la pression P est constante,
[2.4] DH = DE + P DV
[2.5] DH = qP - w + P DV
Comme w = P DV, cela entraîne que DH = qP . Dans ce cas, qP est en fait la variation d’enthalpie à pression constante.
Exemple : C(graphite) + 1/2 O2 (g) ® CO (g)
En ignorant le volume du carbone solide devant ceux des gaz,
DV # 1/2 · 22,4 litres = 11,2 litres par mole de CO formé.
qP = - 110 420 J·mol-1 à 298 K sous la pression de 1 atmosphère.
DE = DH -P DV Comme PV = n RT,
DE = - 110 420 - (-1/2) (1,987 × 4,18 × 298)
DE = - 110 420 + 1238 = - 109183 J·mol-1.
Relation entre qP et qV
Soit un système chimique qui réagit à pression constante - On dit aussi que la réaction se déroule dans des conditions isobares - la variation d’énergie interne au cours de la réaction est
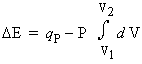
S’il y a variation isotherme de volume, c’est-à-dire s’il y a variation du nombre de moles (nombre de moles de produits - nombre de moles de réactifs = Dn)
|
DE = qP - P(V2 - V1) = qP |
|
Mais, DE = qV car alors dV = 0 : il n’y a pas eu échange d’énergie avec l’extérieur : w = 0.
| [2.6] | qV = qP - Dn R T |
Si Dn = 0, qV » qP
Si Dn ¹ 0, Dn RT = Dn × (2 × 4,18) × 300 = Dn × 2508 J à 300 K
Note : la bombe calorimétrique qui est, et a été, largement utilisée pour mesurer le dégagement d’énergie lors d’une réaction d’oxydation, par exemple, travaille bien entendu à volume constant. Cette technique doit beaucoup à BERTHELOT qui a étudié ce type de réactions vers 1865.
5. L’enthalpie standard de réaction - loi de HESS (1802-1850)
Puisque l’enthalpie, H, ne peut pas être estimée et puisque seule la variation DH peut l’être, on définit arbitrairement une référence, appelée l’enthalpie standard de réaction
![]() .
.
L’état standard d’une substance est représenté par sa forme la plus stable à la pression d’une atmosphère et 298 K. L’enthalpie standard de réaction correspond à la variation d’enthalpie dans les conditions TPN observée au cours de la réaction. Ainsi,
| [a] C(graphite) + O2 (g) ® CO2 (g); | = - 393,13 kJ·mol-1 |
Cette valeur correspond à l'enthalpie standard de formation.
| [b] CO (g) + 1/2 O2 (g) ® CO2 (g); | = - 282,69 kJ·mol-1 |
Cette dernière valeur correspond à l'enthalpie standard de réaction.
Quelle sera l’enthalpie standard de la réaction ?
| [c] C (graphite) + 1/2 O2 (g) ® CO (g); |
|
= ? |
La loi de HESS ou encore le principe de l’état initial et de l’état final, stipule que la variation d’enthalpie d’un système est indépendante du chemin suivi (loi énoncée en 1840).
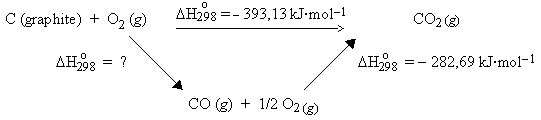
Cette démarche est connue sous le nom de cycle de BORN-HABER. Il s'agit d'imaginer deux successions différentes de réactions ayant en commun leurs points de départ et d'arrivée.
Remarquons qu’en soustrayant l’équation [b] de l’équation [a], on obtient l’équation [c]:
[a] C(graphite) + O2 (g) ® CO2 (g) |
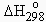 |
= - 393,13 kJ·mol-1 |
[-b] - CO (g) - 1/2 O2 (g) ® - CO2 (g) |
|
= + 282,69 kJ·mol-1 |
______________________________________________________
[c] C (graphite) + 1/2 O2 (g) ® CO (g); |
|
= - 110,44 kJ·mol-1 |
Notons que la loi de HESS est valide pour toute fonction d'état.
6. L’enthalpie standard de formation - DH°f
On peut donc expérimentalement déterminer les enthalpies standards d’un bon nombre de réactions et, de proche en proche, déterminer celles des autres en utilisant la loi de HESS. Cependant, il ne serait pas facile de manipuler des tables de chaleur de plusieurs millions de réactions. Ces tables seraient immenses et interminables. La loi de HESS permet de décomposer une réaction en une suite de réactions selon le chemin :
A- décomposition de tous les réactifs en éléments stables, et
B- reconstitution de tous les produits à partir de ces éléments.
Soit la réaction :
HCOOH (l) ® CO (g) + H2O (l); DHo = ?
Commençons par décomposer l’acide formique en ses éléments stables, puis recomposons ces éléments pour donner les produits:
HCOOH (l) ® H2 (g) + C (graphite) + O2 (g); DHo = DHo(réactifs)
H2 (g) + C (graphite) + O2 (g) ® CO (g) + H2O (l); DHo = DHo(produits).
On définit arbitrairement que la chaleur de formation des éléments les plus stables dans les conditions standards TPN est nulle. Ainsi,
H2 (g) + C (graphite) + O2 (g) ® HCOOH (l)
| [2.7] [2.8] |
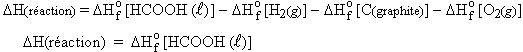 |
et le DHo(réactifs) identifié plus haut est égal à
| [2.9] |
Notons que si la valeur de l’enthalpie de formation standard du graphite est égale zéro,
|
et comme |

|
De la même manière,
H2 (g) + 1/2 O2 (g) ® H2O (l); et C (graphite) + 1/2 O2 (g) ® CO (g); |

|
Construisons le cycle de BORN-HABER suivant :
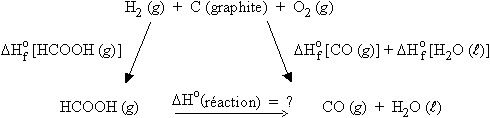
En utilisant la loi de HESS,
| [2.10] [2.11] |
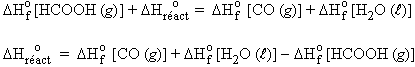
|
Comme cette démonstration est valable pour n’importe laquelle des réactions chimiques,
| [2.12] |
|
Si l’on construit une table des enthalpies standards de formation de chaque produit chimique, on saura calculer l’enthalpie de chaque réaction quelle qu’elle soit. Rappelons seulement que l’enthalpie standard de formation n’est rien d’autre que l’enthalpie de la réaction qui forme le produit à partir des éléments stables. Le nombre de produits étant infiniment moins grand que le nombre de réactions possibles, les tables s’en trouvent allégées d’autant.
Puisque les chaleurs de combustion sont facilement mesurables et que de plus les valeurs des enthalpies de formation de l’eau, du gaz carbonique et de l’oxygène sont connues, il s’ensuit qu’il est facile d’obtenir les chaleurs de formation de n’importe quel combustible. On trouvera donc des tables de chaleurs de combustion. Il en est de même des chaleurs d’hydrogénation.
CmHn + (m + 1/4 n) O2 ® m CO2 + n/2 H2O; DH(comb) = X
avec
| donc, |
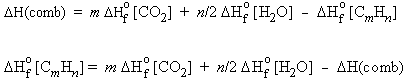 |
Tableau 2.1. L’enthalpie standard de formation de composés à 25 ºC en kJ·mol-1
Substances |
|
Substances |
|
Substances |
|
H2O
( |
- 285,58 |
Ag2O (s) |
- 30,56 |
méthane (g) |
- 74,78 |
H2O (g) |
- 241,60 |
CuO (s) |
-160,93 |
éthane (g) |
- 84,60 |
HCl (g) |
- 92,21 |
FeO (s) |
- 268,77 |
propane (g) |
- 103,75 |
HBr (g) |
- 36,20 |
Fe2O3 (s) |
- 821,37 |
butane (g) |
- 124,61 |
H2S (g) |
- 20,14 |
Fe3O4 (s) |
- 1116,06 |
isobutane (g) |
- 135 |
CO (g) |
- 110,44 |
NaCl (s) |
- 410,60 |
éthylène (g) |
+52,25 |
CO2 (g) |
- 393,13 |
KCl (s) |
- 435,47 |
propylène (g) |
+ 20 |
NH3 (g) |
- 46,15 |
AgCl (s) |
- 126,90 |
1-butène (g) |
- 0,1 |
NO (g) |
+ 90,29 |
NaOH (s) |
- 426,36 |
acétylène (g) |
+ 226,51 |
NO2 (g) |
+ 33,82 |
KOH (s) |
- 426,36 |
propyne (g) |
+185 |
SO2 (g) |
- 296,61 |
Na2SO4 (s) |
- 1381,49 |
benzène (g) |
+83 |
SO3 (g) |
- 394,80 |
CaCO3 (s) |
- 1205,72 |
éthanol
( |
- 277,38 |
7. Influence de la température sur le DHo(réaction) - loi de KIRCHHOFF (1824-1887)
Soit la réaction a A + b B ® m M + n N; DHo(réaction) = ?
Quelle sera la variation de l’enthalpie de la réaction , DHo(réaction), avec une variation de la température ? Écrivons le cycle de BORN-HABER de telle manière que la réaction puisse avoir lieu soit à la température T1, soit à la température T2.
| [a A + b B ] à T2 | [m M + n N ] à T2 | |
| DH' | ¯ DH" | |
| [a A + b B ] à T1 | [m M + n N ] à T1 |
L’application de la loi de HESS conduit à :
[2.13] DHo(T1) = DH' + DHo(T2) + DH"
Définissons la capacité calorifique des réactifs, Cp(réactifs), et des produits, Cp(produits) :
| [2.14] [2.15] |
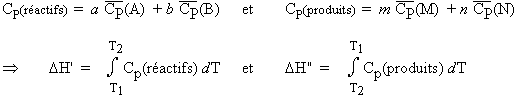 |
[2.16] Þ DHo(T2) = + DHo(T1) - DH' - DH"
| [2.17]
|
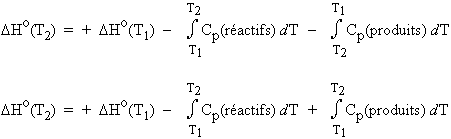 |
On réécrit plus simplement l’expression précédente sous la forme de la loi de KIRCHHOFF, loi énoncée en 1858 :
[2.18] |
|
où
[2.19] DCp = Cp(produits) - Cp(réactifs)
Si la variation des capacités calorifiques est nulle, DCp = 0, l’enthalpie de la réaction est indépendante de la température : DHo(T2) = DHo(T1);
Si au contraire elle n'est pas nulle, DCp ¹ 0, l’enthalpie varie avec la température : DHo(T2) ¹ DHo(T1);
Exemple : Soit la réaction CO + 1/2 O2 ® CO2 avec
|
= - 282,74 kJ·mol-1. |
On donne aussi:
|
(CO) = + 29,13 J·degré-1·mol-1, |
|
(O2) = + 29,13 J·degré-1·mol-1, |
|
(CO2) = + 29,13 J·degré-1·mol-1, |
Calculer la valeur de l'enthalpie de la réaction à 100 °C (398 K) .
La variation des capacités calorifiques à pression constante DCp doit d'abord être calculée :
DCp = 37,45 - 1/2 (29,47) - 29,13 = - 6,415 J·degré-1·mol-1.
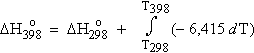 |
|||
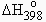 |
= |
|
- 6,415 (398 - 298) |
|
= | - 282 740 - 641,5 = - 283 381 J·mol-1. | |
Dans cet exemple, on a considéré que les capacités calorifiques sont des quantités fixes: elles ne variaient pas avec la température. Cette approximation est valable lorsque l’intervalle de température considéré est relativement étroit. Ce n’est plus le cas lorsque cet intervalle DT, est supérieur à 100 o.
8. L’enthalpie de liaison
Il est bien évident qu’un système réactionnel implique la rupture de certaines liaisons et la reconstruction de nouvelles. Il est tentant d’essayer de calculer les enthalpies requises ou les énergies requises pour briser ou reconstruire ces liaisons chimiques. Par exemple :
| CH4 (g) | ® C (g) | + 4 H· (g) | |
|
- 74,85 | + 718,38 | + 4 (217,94) kJ·mol-1. |
DH°(réaction) |
= + 718,38 + 4 (217,94) - (- 74,85) = 1 665 kJ·mol-1. |
Comme la réaction consiste essentiellement en la construction de quatre liaisons C-H de type s, on peut estimer que l’énergie requise pour briser une telle liaison est 416,25 kJ·mol-1 soit encore 99,6 kcal·mol-1. On peut ainsi, de proche en proche établir une liste, une table des énergies de liaisons courantes (Tableau 2.2). On trouve dans la littérature thermodynamique des tables de chaleurs de combustion. Les réactions impliquées étant relativement semblables, et l’exothermicité des réactions relativement importantes on procède avec l’instrument appelé bombe calorimétrique. Cet appareil maintient le volume constant et permet de mesurer la chaleur dégagée pendant la réaction (voir Fig. 2.1).
Tableau 2.2. Enthalpies de quelques liaisons simples (kJ·mol-1) à 25 ºC
Br |
C |
Cl |
F |
H |
I |
N |
O |
S |
P |
|
Br |
193 |
285 |
219 |
249 |
366 |
178 |
234 |
218 |
264 |
|
C |
348 |
339 |
489 |
413 |
218 |
305 |
358 |
272 |
264 |
|
Cl |
242 |
253 |
431 |
211 |
192 |
208 |
271 |
322 |
||
F |
159 |
567 |
280 |
278 |
193 |
327 |
503 |
|||
H |
436 |
298 |
391 |
463 |
367 |
322 |
||||
I |
151 |
234 |
184 |
|||||||
N |
163 |
201 |
||||||||
O |
146 |
335 |
||||||||
S |
255 |
|||||||||
Voir Aylward, G. H. et T. J. V. Findlay, SI Chemical Data, J. Wiley & Sons, 2e éd., (1974). |
||||||||||
Note : ces enthalpies de liaison correspondent à l’énergie qu’il faut fournir pour rompre chacune d’entre elles.
Tableau 2.3. Enthalpies de quelques liaisons multiples (kJ·mol-1) à 25 ºC
Liaisons doubles |
Liaisons triples |
|||||
C |
N |
O |
S |
C |
N |
|
C |
614 |
615 |
745 |
536 |
839 |
891 |
N |
418 |
607 |
945 |
|||
O |
498 |
|||||
Voir Aylward, G. H. et T.J.V. Findlay, SI Chemical Data, J. Wiley & Sons, 2e éd., (1974). |
||||||
On trouvera d’autres énergies de liaison dans : Bond dissociation energies in simple molecules, NSRDS-NBS 31, U.S. Dept of Commerce, janvier 1970.
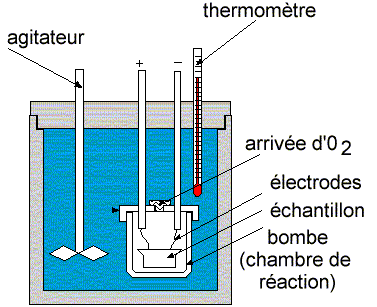
Figure2.1. Bombe calorimétrique.
Compte tenu de l’équivalence ou de la relation très étroite entre les processus chimiques mis en cause et les variations de l’enthalpie, on conçoit aisément, par exemple, que la chaleur des réactions d’hydrogénation des alcènes ne doit pas dépendre de l’alcène impliqué. En effet la réaction d’hydrogénation du propène en propane, du 1-butène en butane normal, ... met en cause, dans chaque cas, l’ouverture ou le bris de la liaison p de l’alcène, la rupture de la liaison s de la molécule d’hydrogène et la formation de deux liaisons s de type C-H. On observe en effet que la chaleur de cette réaction est de l’ordre de -122 ± 3 kJ·mol-1 dans le cas des alcènes linéaires avec double liaison en position terminale. La valeur est plus élevée dans le cas de l’éthylène : DH = -137 kJ·mol-1 . Cette différence est due au fait que la répartition spatiale des orbitales est ici légèrement différente ou encore que l’encombrement électronique autour de la double liaison est différent de celui observé dans le cas des oléfines terminales.
CH3-(CH2)n-CH=CH2 + H2 ® CH3-(CH2)n+1-CH3 avec n = 0, 1, 2,...
Cette concordance thermochimique se traduit aussi de la même manière et pour les mêmes raisons en cinétique. Par exemple, la vitesse de la réaction d’acétylation des alcools terpéniques catalysée par la N,N-diméthyl-4-aminopyridine dans le tétrahydrofuran à 25 ºC est indépendante de la nature de l’alcool : tableau suivant. Tout au plus, les valeurs indiquées laissent-elles présager un effet stérique important lorsque l’on passe des alcools primaires aux alcools secondaires.
Tableau 2.4. Acétylation
de quelques alcools :
Constantes de vitesse de pseudo premier ordre (min-1)
alcools primaires |
||||||
1-pentanol |
citronellol |
cis-3-hexén-1-ol |
2-phényléthanol |
géraniol |
nérol |
myrténol |
6,2 10-2 |
6,9 10-2 |
5,6 10-2 |
7,7 10-2 |
8,9 10-2 |
3,9 10-2 |
8,2 10-2 |
alcools secondaires |
||||||
cyclohexanol |
1-menthol |
thimbérol |
isobornéol |
bornéol |
||
1,25 10-2 |
1,6 10-2 |
1,7 10-2 |
0,13 10-2 |
0,84 10-2 |
||
| Lamaty, G. et al., Parfums, Cosmétiques, Arômes, 101, 53 (octobre - novembre 1991). | ||||||
9. Influence de la pression sur le DHo(réaction)
Pour un gaz parfait, la relation entre l'enthalpie et la pression est donnée par la relation [2.3] :
![]()
Les fonctions enthalpie H et énergie E sont des fonctions d'état.
- Pour un gaz idéal, le produit PV est constant à température constante. Donc:
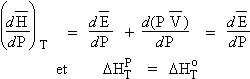
Pour un gaz idéal, la valeur l’enthalpie standard de réaction est indépendante de la pression.
- Pour un gaz réel, pour un solide ou un liquide, ce n’est plus vrai. On montre que la variation d'enthalpie avec la pression est donnée par la relation :
|
|
est la chaleur molaire de variation de pression (ce n’est pas une chaleur molaire de changement de phase). On montrera plus loin que cette valeur est reliée à a le coefficient de dilatation thermique : |
|
[2.24] |
 |
[2.25] |
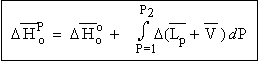 |
- Si le constituant est un gaz réel idéal :

Donc,
1 - a T = 1 - RT / PV = 0
car PV = RT. Cela n’est bien sûr plus vrai si le gaz obéit à l’équation d’état de VAN DER WAALS :
| [2.26] |
|
- Dans le cas des solides et des liquides, le volume molaire est approximativement constant puisque ces phases condensées sont réputées incompressibles.
|
|
Dans cette expression, la pression P est exprimée en atmosphères.
| 10. Conclusions
On ne sait pas a priori déterminer l’énergie interne d’un système, même fermé. On sait par contre en mesurer les variations au cours d’une transformation physico-chimique en particulier lorsque la transformation se fait à volume constant (cas moins fréquent). Lorsque les transformations se font à pression constante on définit la fonction enthalpie. Cette fonction tient compte de l’échange de travail avec l’extérieur du système. Les variations d’énergie interne et d’enthalpie sont reliées entre elles à travers une relation qui implique la variation du nombre de moles observée au cours de la réaction. Les variations d’énergie interne et d’enthalpie avec la températures sont mathématiquement reliées aux capacités calorifiques à volume ou à pression constante.
|
11.1 La chaleur (l’enthalpie) de combustion d’une mole d’isobutane dans un excès d’oxygène à volume constant (bombe calorimétrique) est de -2871,65 kJ·mol-1 avec formation concomitante de CO2 (g) et de H2O (l). Calculez la valeur de la chaleur de formation de l’isobutane.
11.2 La combustion d’une mole de benzène, en présence d’un excès d’oxygène et à volume constant, produit un dégagement de chaleur de 3301,51 kJ·mol-1. Calculez l’enthalpie de formation de ce composé.
Réponse : 85,99 kJ/mol
11.3 À partir des données suivantes, calculez l’enthalpie standard de formation du carbure d’aluminium solide (Al4C3) :
Composé |
CO (g) |
CO2 (g) |
Al2O3 (s) |
|
- 110,35 |
- 393,13 |
- 1672 |
On sait en outre que la réaction suivante est exothermique :
Al4C3 (s) + 9 CO2 (g) ® 2 Al2O3 (s) + 12 CO (g);
DH°(réaction) = - 935,1 kJ·mol-1.
Réponse :
-194,99 kJ/mol
11.4 Les capacités calorifiques molaires à pression constantes exprimées en J·(K·mol)-1 de l’eau, de l’oxyde de carbone, du gaz carbonique et de l’hydrogène sont les suivantes :
| Molécules | 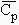 |
= a' + b' T + c' T2 | |
H2O (g) |
+ 36,83 |
- 7,94 10-3 T |
+ 9,28 10-6 T2 |
CO (g) |
+ 27,17 |
+ 4,18 10-3 T |
|
H2 (g) |
+ 27,17 |
+ 3,76 10-3 T |
|
CO2 (g) |
+ 29,26 |
+ 29,68 10-3 T |
- 7,77 10-6 T2 |
On étudie la réaction de synthèse du gaz à l’eau :
H2 (g) + CO2 (g) ® H2O (g) + CO (g)
Calculez :
1- La chaleur de réaction dans les conditions standards TPN.
2- Les variations d’enthalpie de la réaction à 400, 500, 600, 700, et 800 K.
11.5 On étudie la réaction de réduction du gaz carbonique en oxyde de carbone gazeux à l’aide du carbone graphite. En utilisant les données numériques apparaissant dans les tableaux 2.1 et 2.7b, établissez la fonction
DH°(réaction) = ƒ(T).
Calculez les valeurs numériques de cette fonction entre 300 et 900 K par valeur de 100 K. Tracez la courbe entre ces mêmes valeurs. Établissez graphiquement, numériquement et analytiquement s’il y en a un, le maximum que peut prendre cette fonction.
C (graphite) + CO2 (g) ® 2 CO (g)
11.6 À l’aide du principe de l’état initial et de l’état final, calculez l’enthalpie de formation de AgCl (s) à 25 ºC. On donne :
Ag2O (s) + 2 HCl (g) ® 2 AgCl (s) + H2O (l); DHo = - 324,41 kJ
2 Ag (s) + 1/2 O2 (g) ® Ag2O (s); DHo = - 30,56 kJ
1/2 H2 (g) + 1/2 Cl2 (g) ® HCl (g); DHo = - 92,21 kJ
H2 (g) + 1/2 O2 (g) ® H2O (l); DHo = - 285,58 kJ
Réponse : DHof(AgCl(s)) = -126,9 kJ
11.7 Le fer existe dans les conditions standards avec une structure cristalline appelée a; il est de plus magnétique. À 760 ºC, il perd cette propriété: il devient paramagnétique. À 910 ºC, le fer change de structure cristalline et devient le fer g.
| [1] Fe (a, magnétique) [2] Fe (a) |
Fe (a, paramagnétique) ;
DH = 2 759
J·mol-1 Fe (g) ; DH = 920 J·mol-1 |
a) L’enthalpie de formation standard de FeO (s) est de - 264,2 kJ·mol-1, calculez la chaleur de réaction standard de la réaction suivante :
[3] Fe (a) + 1/2 O2 (g) ® FeO (s)
Réponse : -264,2 kJ/mole
| Composé |
|
= a + b T + c T-2 | |
Fe, a, magn. |
+ 17,5 |
+ 24,75 10-3 T |
|
FeO, s |
+ 48,74 |
+ 8,36 10-3 T |
- 2,80 10+5 T-2 |
O2, g |
+ 29,93 |
+ 4,18 10-3 T |
- 1,67 10+5 T-2 |
c) Calculez la chaleur de la réaction suivante à 760 ºC.
[4] Fe (a, param.) + 1/2 O2 (g) ® FeO (s)
d) La capacité calorifique du fer paramagnétique ne dépend pas de la température :
|
= 37,62 J·(K·mol)-1. |
Calculez la chaleur de la réaction [4] à 910 ºC.
e) Calculez la chaleur de la réaction suivante à 910 ºC :
| [5] Fe (g) + 1/2 O2 (g) ® FeO (s) |
f) La capacité calorifique du fer (g) est connue :
|
= 7,69 + 19,48 10-3 T J·(K·mol)-1 |
Calculez la chaleur de la réaction [5] à 1350 ºC.
11.8. L’oxyde de chrome pur, Cr2O3, réagit avec l’aluminium pur à 25 °C pour donner l’alumine et le chrome liquide pur. Étant donné que la température finale atteinte est de 1900 °C, calculer la chaleur dégagée et absorbée par le milieu extérieur par kilogramme d’aluminium. Les données disponibles sont :
|
Tfus |
DH°(fusion) |
|
|
Al (s) Cr (s) Cr ( l)Al2O3 (s)Cr2O3 (s) |
0 0 - - 1672 - 1128 |
679 1850 - - - |
10,45 19,23 - - - |
- 24,41 + 9,86 10-3 T - 3,68 105 T-2 39,29 114,45 + 12,87 10-3 T - 34,28 105 T-2 - |
* : jusqu’à 1800 °C (2073 K) |
||||
Réponse : 23,71 % en masse d’aluminium et 76,29 % en masse d’oxyde de fer.
Calculez la quantité d’énergie libérée à 25 °C lors de la réaction complète. En déduire la quantité d’énergie fournie par gramme d’aluminium.
Réponse : -15,48 kJ/g d’aluminium
Dans un creuset contenant 27,2 kg de mélange stœchiométrique, la température s’élève à 2600 °C. Évaluer la chaleur spécifique apparente du creuset. Cette température étant excessive. On ajoute au mélange une certaine quantité de limaille de fer. La température finale est de l’ordre de 2400 °C. En considérant que la capacité calorifique du creuset est constante entre 300 et 2800 K, calculez la quantité de limaille de fer ajouté à 27,2 kg de mélange stœchiométrique. Les données disponibles sont :
(kJ/mole) |
Ttransf | DH°(fusion) (kJ/mole) |
(J · mol-1 · °C-1) |
|
| Fe (a, magn.) | 0 | 760 (® a, non magn.) | 2,75 | 17,5 + 24,75 10-3 T |
| Fe (a, non magn.) | - | 910 (® g) | 0,92 | 37,62 |
| Fe (g) | - | 1401 (® d) | 0,88 | 7,69 19,5 10-3 T |
| Fe (d) | - | 1539 (® liquide) | 15,5 | 43,9 |
| Fe (liquide) | - | - | - | 41,8 |
| Al (s) | 0 | - | - | - |
| Al2O3 (s) | - 1 672 | - | - | voir tableau précédent |
| Fe3O4 (s) | - 1 155,66 | - | - | - |
Réponse : 3,60 kg de fer.
11.9 À partir des données apparaissant dans le Tableau 2.2, calculez l’enthalpie standard de la réaction :
CH4 (g) + Cl2 (g) ® HCl (g) + CH3Cl (g).
Réponse : -115 kJ/mole
11. 10. Température de flamme
Calculez la température maximum des produits de combustion de la flamme issue d’un mélange stœchiométrique air – méthane lui-même à la température de 300 K et à la pression atmosphérique. On supposera que 90 % du méthane sont effectivement oxydés.
CH4 + 2 O2 ® CO2 + 2 H2O ; DH298 = 756,3 kJ
On donne les valeurs des capacités calorifiques suivantes (en J · K-1 · mol-1 entre 300 et 2000 °C) :
CP(CH4, g) = 31,5 + 0,014 T
CP(O2, g) = 26,0 + 0,0045 T
CP(N2, g) = 28,3 + 0,003 T
CP(CO2, g) = 32,35 + 0,005 T
CP(H2O, g) = 34,2 + 0,002 T
Réponse : T = 2 225 K = 1 952 °C
Pour aller plus loin :
Dernière mise à jour : 2021-07-08.
x x x x x x x x x x x x x x x
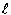 )
)