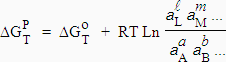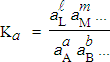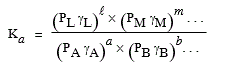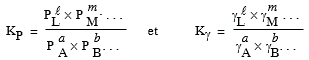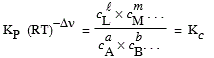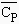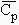CHAPITRE VII
ÉQUILIBRES CHIMIQUES
|
Après avoir bien compris les propriétés des principales fonctions thermodynamiques et leurs applications aux corps purs, on peut maintenant envisager l’application de ces fonctions aux réactions chimiques. Questions : Objectifs : |
Soit le mélange de méthanol et d’acide acétique. Laissé à lui-même, il réagit et se transforme partiellement à la température normale en acétate de méthyle et en eau. Si l’on mélange au contraire de l’acétate de méthyle et de l’eau, le système va évoluer vers la formation de méthanol et d’acide acétique. Si, en outre, on a pris soin dans chacun des deux cas de réaliser des mélanges équimoléculaires, on obtient le même mélange final.
CH3OH + CH3COOH ![]() CH3COOCH3
+ H2O
CH3COOCH3
+ H2O
Les deux systèmes précédents évoluent l’un vers l’autre et atteindront un état d’équilibre dynamique. Loin d’être figés, les réactifs continuent de réagir. Il y a donc réactions directe (de gauche à droite) et indirecte (de droite à gauche) de façon ininterrompue. On dit aussi qu’il y a équilibre entre deux réactions. Les vitesses des deux réactions étant identiques, on obtient en apparence un état de non réaction.
Définitions
On appelle mélange tout système constitué d’au moins deux corps purs. Cette définition ne présume pas que les corps purs se dissolvent ou non mutuellement l’un dans l’autre. Par exemple, si la mixtion de l’eau et du pentane forme un mélange, il en est de même du chlorure de sodium, NaCl, immergé et donc solubilisé dans de l’eau.
On appelle équilibre homogène tout mélange qui résulte en la formation d’une seule phase solide, liquide ou gazeuse. Cette phase est homogène : ses propriétés sont identiques quel que soit le lieu considéré à l’intérieur de cette phase. Par exemple, a-pinène + b-pinène constitue une phase homogène organique. Comme les réactifs ne réagissent pas entre eux, il n’y pas d’équilibre chimique, mais seulement un équilibre physique. Le mélange de nitrate de potassium et de chlorure de sodium en solution aqueuse conduit à un équilibre chimique :
NaCl + KNO3
![]() KCl + NaNO3
KCl + NaNO3
On appelle équilibre hétérogène tout mélange qui conduit à la formation d’un équilibre chimique faisant intervenir au moins deux phases homogènes. La dissolution d’iode dans le mélange eau - tétrachlorure de carbone conduit à une distribution d’iode entre les deux solvants. Il y a équilibre physique. Par contre, le mélange carbone graphite + bioxyde de carbone gazeux forme l’équilibre chimique hétérogène suivant :
C (graphite) + CO2 (g) ![]() 2 CO (g)
2 CO (g)
2. Rappels - La règle des phases
2.a Définitions
a- phase j : Une phase est toute partie homogène d’un système physiquement distincte des autres parties dont elle est séparée par des surfaces de délimitation bien définies.
b- nombre de composants C :
Chaque phase contient un nombre d’espèces bien défini. Le nombre de composants est ce nombre d’espèces diminué du nombre de relations chimiques qui les relient.
Nota : ne pas confondre composant et composé!
c- variance u : Lorsque le nombre de composants d’un système et les composants de chacune des phases sont précisés, c’est le nombre de facteurs qu’il faut préciser pour que l’équilibre soit défini.
Nota : la variance est aussi appelée le nombre de degré de liberté du système.
2.b Calcul théorique de la variance
a- nombre de facteurs :
i- Température et pression : 2
ii- S’il y a C composants dans une phase, il a C - 1 concentrations indépendantes puisque SiCi = 1
Si j est le nombre de phases du système, il y aura j(C - 1) concentrations indépendantes.
b- nombre de relations :
i- Pour un composant il y a j - 1 relations thermodynamiques qui relient les quantités partielles molales entre les j phases. Comme il y a C composants, il y aura C (j - 1) relations thermodynamiques (voir tableau 7.1).
c- variance
La variance u est égale au nombre de facteurs diminué du nombre de relations :
u = 2 + j(C - 1) - C (j - 1)
| [7.1] Þ |
|
Tableau 7.1. Représentation schématique d’un système de n composés
en équilibre
dans j phases
Phase 1 |
Phase 2 |
Phase 3 |
Phase j |
Phase j |
||||
[A]1 |
|
[A]2 |
|
[A]3 |
|
[A]j |
|
[A]f |
[B]1 |
|
[B]2 |
|
[B]3 |
|
[B]j |
|
[B]f |
[C]1 |
|
[C]2 |
|
[C]3 |
|
[C]j |
|
[C]f |
[D]1 |
|
[D]2 |
|
[D]3 |
|
[D]j |
|
[D]f |
[I]1 |
|
[I]2 |
|
[I]3 |
|
[I]j |
|
[I]f |
[N]1 |
|
[N]2 |
|
[N]3 |
|
[N]j |
|
[N]f |
Dans le cas où la phase gazeuse n’intervient pas, le paramètre pression n’intervient pas puisque les solides et les liquides sont réputés incompressibles :
u = C + 1 - j
C’est le cas des courbes de fusion, des transformations allotropiques.
3. Constantes thermodynamiques à l’équilibre ou d’équilibre
Soit la
réaction a A + b
B 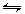 L + m M
L + m M
| [7.2] |
|
Dans cette équation, aA est l’activité du réactif A,...
Pour qu’il y ait équilibre, il faut que la variation d’énergie libre au cours de la réaction soit nulle :
|
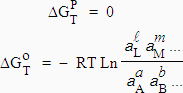 |
À température donnée, la valeur du membre de droite de l’équation [7.3] est constante.
[7.4] Þ |
|
Ka est la constante thermodynamique à l’équilibre et
| [7.5] |
|
à l’équilibre. Il faut noter que
Ka (réaction directe) = 1 / Ka (réaction inverse)
4. Introduction des mesures KP, Kc, Ky et Kg
4.a Relation entre Ka, KP et Kg
[7.6] Dans un gaz, a = P g où
- a est l’activité ou encore appelé la fugacité,
- P est la pression partielle, et
- g est le coefficient d’activité
L’activité d’un constituant dans un gaz mesure en quelque sorte son influence réelle au sein du mélange gazeux, Si le mélange est idéal, l’influence réelle de chaque gaz est égale à sa fraction molaire. On remarque que cette idéalité s’amenuise au fur et à mesure que le mélange s’approche des conditions de liquéfaction ou encore que la pression totale s’accroît comme le montre le tableau suivant.
Tableau 7.2. Relation entre la pression et l’activité pour quelques gaz
P (atm) |
Hydrogène (100 °C) |
Ammoniac (200 °C) |
Gaz carbonique (60 °C) |
|||
a (atm) |
g |
a (atm) |
g |
a (atm) |
g |
|
0 25 50 100 200 300 400 500 |
0,0 25,3 51,5 105 222 351 492 650 |
1,00 1,01 1,03 1,05 1,11 1,17 1,23 1,30 |
0,0 23,9 45,7 84,8 144 193
|
1,000 0,954 0,913 0,848 0,720 0,642
|
0,0 23,2 42,8 70,4 91,0 112
|
1,000 0,928 0,856 0,704 0,455 0,373
|
Extrait de Maron, S. H. & C. F. Prutton, Principles of Physical Chemistry, McMillan Comp., Ney York, 1965. |
||||||
Loi de LEWIS :
L’activité d’un composé faisant partie d’un mélange gazeux est égale à la pression partielle de ce composé gazeux multipliée par le coefficient d’activité du gaz à la pression totale du mélange. |
[7.7] ai = Pi gi = xi P gi où
- xi est la fraction molaire du gaz i,
- P est la pression totale, et
- gi est le coefficient d’activité.
Þ |
|
NOTE: de l’équation [7.7] à la fin de ce chapitre, on a implicitement supposé que la pression de référence est la pression atmosphérique. Si ce n’est pas le cas, chaque valeur de P, Pi, PL, … doit être remplacée par le rapport P/P0, Pi/P0, PL/P0,
[7.8] Þ Ka = KP Kg
[7.9] où |
|
Dans cette équation, Ka est la constante d’équilibre mesurée ou exprimée en fonction des activités de chacun des constituants de l’équilibre. KP est la constante d’équilibre mesurée ou exprimée en fonction des pressions partielles. Kg est la constante d’équilibre mesurée ou exprimée en fonction des coefficients d’activité. Pour un gaz réel, cette valeur dépend de la pression totale. Pour un gaz parfait : Kg = 1 et Ka = KP. En général, pour un gaz réel, Kg = 1 tant et aussi longtemps que la pression totale reste inférieure à 10 atmosphères.
4.b Expression de KP et de KC
En phase gazeuse, selon la loi de MARIOTTE,
[7.10] P = (n/V) R T = c R T où c représente le nombre de moles par unité de volume.
[7.11] Þ
|
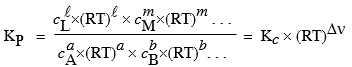 |
Dans cette équation, regroupons les concentrations:
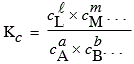
et Dn est la variation de molécularité de la réaction.
Dn
= ![]() + m + ..
-
a
- b
- . .
+ m + ..
-
a
- b
- . .
[7.12] KP = Kc (R T)Dn
Si Dn = 0, Þ (R T)Dn = 1 et KP = Kc ,
la transformation se fait à volume constant.
4.c Expression de KP en fonction de Kg
( y représente la fraction molaire) :
[7.13] |
|
L'équation [7.12] peut se réécrire sous la forme :
[7.14] Þ |
|
Comme cx = nx/V , yx = nx/n et PV = n R T
yx = cx (V/n) = cx (RT / P)
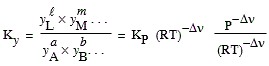
[7.15] Ky = KP P-Dn
Si à nouveau, la variation de molécularité de la réaction est nulle, Dn = 0, Þ Ky = KP puisque P-Dn = 1.
5. La loi de LE CHATELIER - BRAUN ou encore le principe de LE CHATELIER
Considérons un système à deux températures différentes T et T',
ou encore
![]()
Þ
|
 |
Supposons que les températures T et T' soient proches de telle manière que les enthalpies et les entropies de réaction ne changent pas :
|
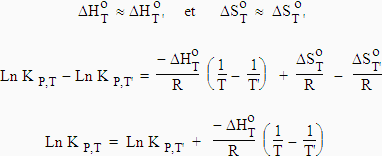 |
1- Supposons que la réaction soit exothermique : la variation d’enthalpie est négative, DH < 0 et que T ' > T
Þ Ln KP,T > Ln KP,T'
Autrement dit, la constante d’équilibre décroît lorsque la température T croît : l’équilibre se déplace vers la gauche quand la température croît.
2- Supposons que la réaction soit endothermique : la variation d’enthalpie est positive DH > 0 et que T ' > T
Þ Ln KP,T < Ln KP,T'
Autrement dit, la constante d’équilibre croît lorsque la température T croît : l’équilibre se déplace vers la droite quand la température croît.
Dans chaque cas, pour contrer l’élévation de température, le système tend à absorber de l’énergie. Si l’on applique un stress à un système en équilibre, celui-ci réagit de manière à éliminer ce stress, de façon à annihiler la déformation imposée. Ce principe est aussi aisément vérifiable dans le cas où la déformation appliquée est une variation de pression (voir aussi LE CHATELIER).
a) À haute pression, l’ajout d’un gaz inerte modifie les coefficients d’activité gi et donc la valeur de la constante d’équilibre Kg.
b) À faible pression, il n’y a pas d’effet de cette nature (ou celui-ci est marginal). Il y a cependant changement des pressions partielles tout comme de la pression totale.
6.a Si le système opère à volume constant
Écrivons l’expression de la constante KP :
| [7.17] |
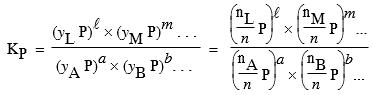 |
car yL = nL / n et n = nA + nB + ... + nL + nM + .... Donc,
| [7.18] [7.19] |
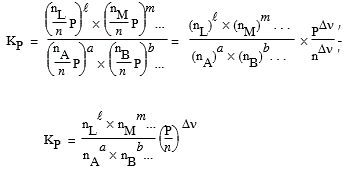 |
Si le volume V est constant, quand le nombre n croît, la pression P croît également et n est proportionnel à P : n a P. Il en résulte que le rapport P/n est constant quel que soit n. Il n’y a donc pas d’effet.
Þ Dans un système opérant à volume constant (système isochore) l’ajout d’un gaz inerte n’ a pas d’effet sur la réaction.
6.b Si le système opère à pression constante
Dans le cas de système isobare, trois cas se présentent.
6.2-a Si la molécularité ne change pas au cours de la réaction, si Dn = 0, lorsqu’on ajoute un gaz inerte, le rapport
(P/n)Dn = 1 et il n’y a pas d’effet sur la réaction.
6.2-b Si Dn > 0, quand n croît, le précédent rapport décroît. Pour compenser cette décroissance et afin de respecter la constant d’équilibre, le rapport
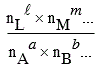
doit croître. Il doit donc y avoir déplacement d’équilibre vers la droite et donc une augmentation dans la formation des produits.
6.2-c Si Dn < 0, quand n croît, le rapport (P/n)Dn croît.
Pour compenser cette croissance et afin de respecter la constante d’équilibre, le rapport
doit décroître. Il doit donc y avoir déplacement d’équilibre vers la gauche. En d’autres termes, il y a diminution dans la formation des produits.
7. Constante d’équilibre pour une réaction hétérogène
Ces réactions sont industriellement nombreuses et économiquement importantes. Elles incluent les réactions de décomposition d’oxydes, de carbonates, d’hydrates salins, ...
Soit la réaction
ZnO (s) + H2 (g) ® Zn (s) + H2O (g)
l’équation [7.3] s’écrit
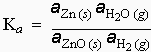
L’activité d’un solide pur ou d’un liquide pur est égale à l’unité jusqu’à de hautes pressions et pratiquement à toutes températures.
Þ |
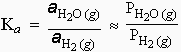 |
On peut donc ignorer les solides et les liquides purs dans l’établissement de la constante d’équilibre.
Démonstration thermodynamique :
Soit la réaction
a A + b B + . . .
®
![]() L + m
M + . . .
L + m
M + . . .
et supposons que les constituant A, B... L, M... se partagent en deux groupes :
a- des solides ou liquides purs distingués par l’indice i, et
b- des gaz parfaits distingués par l’indice j.
| [7.20]
|
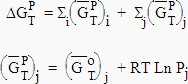 |
Pour un solide ou un liquide pur, la variation de l’énergie libre avec la pression est négligeable. Donc,
| [7.21]
|
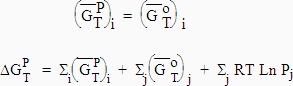 |
À l’équilibre, la variation de l'énergie libre est nulle :
![]()
| [7.23] Þ |
|
Conclusion :
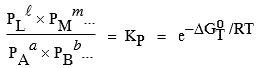
Dans l’expression de la constante
KP, on ne tient compte que des pressions partielles des composés gazeux. Par contre, dans
l’expression de
![]() , tous les composants interviennent.
, tous les composants interviennent.
Exemple 1, oxydation du fer par l’eau. L’équilibre suivant gouverne le système :
3 Fe (s) + 4 H2O (g) ![]() Fe3O4
(s) + 4 H2 (g)
Fe3O4
(s) + 4 H2 (g)
|
avec |
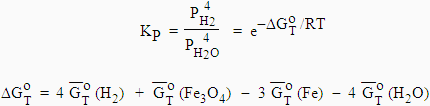 |
On aurait pu écrire :
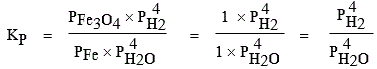
Il aurait alors fallu comprendre que dans la phase solide Fer, il n’y a que du fer et que sa pression hypothétique est de un (1) atmosphère ou encore, ce qui est plus juste, que son activité thermodynamique est égale à l’unité.
Exemple 2, carbonatation de la chaux vive :
Soit la réaction CaO (s)
+ CO2 (g) ®
CaCO3 (s)
Au-dessus de la chaux vive, CaO, ajoutons graduellement du gaz carbonique. Tant et aussi longtemps que l’on n’a pas atteint la constante d’équilibre de décomposition du carbonate de calcium, PCO2, il ne se passe aucune réaction chimique. Dès que cette pression est atteinte, la réaction de carbonatation démarre et la pression de CO2 demeure constante jusqu’au moment où toute la chaux vive a été transformée en carbonate de calcium. Pendant cette étape de carbonatation appliquons la règle de la variance :
u = C + 2 - j = 2 (3 constituants - 1 réaction chimique) + 2 (T et P) - 3 phases = 1.
Comme les deux solides ne sont pas solubles l’un dans l’autre, il faut compter trois phases. Si la réaction de carbonatation se fait à la température ordinaire, donc à une température donnée, la pression de gaz carbonique est fixée par le système. Elle l’est à travers la détermination de la constante d’équilibre de la réaction. Lorsque la chaux vive a disparu, le nombre de phases est réduit à deux, le système redevient bivariant et la pression de gaz carbonique peut augmenter.
Exemple 3, hydratation du sulfate de cuivre
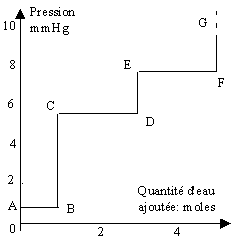
Le système eau-sulfate de cuivre est soumis aux trois équilibres suivants :
[1] CuSO4,H2O ® CuSO4 + H2O; KP = 0,8 mmHg,
[2] CuSO4,3 H2O ® CuSO4,H2O + 2 H2O; KP = (5,6 mmHg)2,
[3] CuSO4,5 H2O ® CuSO4,3 H2O + 2 H2O; KP = (7,8 mmHg)2.
|
|
Figure 7.1. Variation de la pression durant l’hydratation
d’une mole de sulfate de cuivre anhydre.
On observera donc dans ce cas trois paliers de pression (Fig. 7.1). Ces paliers sont bien sûr déterminés par les constantes d’équilibre des réactions. En ajoutant, à 25 ºC, de la vapeur d’eau au-dessus du sulfate de cuivre anhydre, on atteint tout d’abord un plateau situé à 0,8 mmHg. La pression demeure stable jusqu’à transformation du sulfate anhydre en sulfate monohydraté. Puis la pression s’élève à nouveau jusque 5,6 mmHg où elle demeure stable jusqu’à transformation complète en sulfate dihydraté. On observe enfin un autre plateau de pression à 7,8 mmHg jusqu’à transformation complète en sulfate pentahydraté.
8. Variation de KP et de Ka avec T - Équation de VAN'T HOFF
À l’équilibre, l’équation [7.5] s'écrit,
| - RT Ln Ka |
| [7.24] Þ |
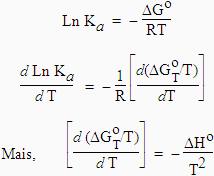 |
voir l’équation [4.4] |
| [7.25] Þ |
|
Dans le cas d’un gaz parfait, Ka = KP
| [7.26] |
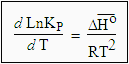 |
C’est l’équation de VAN'T HOFF (1884). |
Pour intégrer cette équation, il faut non seulement connaître la variation de l’enthalpie standard de réaction, DHo, mais également les variations de DHo avec la température : DHo = ƒ(T). En considérant l’intervalle de température assez petit, on peut supposer que DHo est constant.
[7.27]
[7.28] Þ
[7.29] |
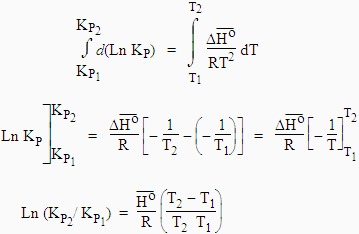 |
Ou encore de façon plus générale, en introduisant les valeurs relatives à la température T1 dans la constante d’intégration, on obtient :
| [7.30] [7.31] |
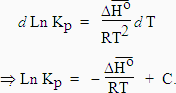 |
Sur un graphe Ln Kp = ƒ(1/T), la pente sera égale à
-![]() /R et C est
l’ordonnée à l’origine. Si
/R et C est
l’ordonnée à l’origine. Si ![]() varie sur l’intervalle de température. . . Les tables donnent les variations de
capacité calorifique à pression constante, DCp,
avec la température : voir Chapitre
I.3. On obtient des formules de la forme
varie sur l’intervalle de température. . . Les tables donnent les variations de
capacité calorifique à pression constante, DCp,
avec la température : voir Chapitre
I.3. On obtient des formules de la forme
| et |
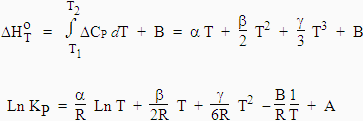 |
Si la température T = 0,
|
|
= B. |
Ceci n’est vrai qu’au zéro absolu où cette valeur est purement hypothétique.
La constante B ne
peut être confondu avec la
variation d’enthalpie standard 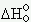 .
.
[7.32] |
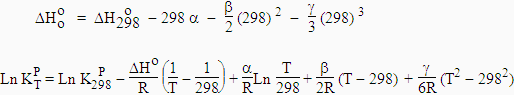 |
Si DHo est constant sur l’intervalle de température, on retrouve l’expression [7.29].
9. Autre forme de l’équation de VAN'T HOFF
On se souvient de l’équation [7.12],
[7.12] KP = Kc (RT)Dn
De plus, DH = DE + PDV avec PDV = Dn RT car (PV = n RT)
Ln KC,T = Ln KP,T - Dn Ln RT
| [7.33]
[7.34]
[7.35] |
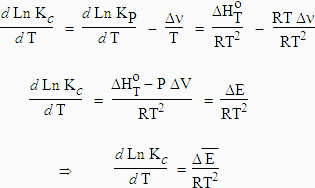 |
La constante d'équilibre par rapport aux concentrations, Kc, joue par rapport à l'énergie interne le même rôle que la constante d'équilibre KP par rapport à l'enthalpie (voir équation [7.26]).
10. Équilibres ne faisant intervenir que des phases condensées pures
(sans phase gazeuse, ni solutions liquides ou solides)
| Dans ces cas, | 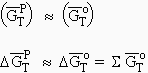
|
|
et quelle que soit la pression : |
La condition d’équilibre se réduit à
![]()
Lorsqu’il y avait un gaz, il y avait possibilités d’équilibre à toute température avec P = ƒ(T). Dans le cas où le système ne fait intervenir que des phases condensées pures, il n’existe qu’une seule température (si cette température existe) à laquelle le
![]()
s’annule. À toute autre température, il y aura réaction complète, dans un sens ou l’autre.
Fe (s) + S (s) ® FeS (s)
| 11. Conclusions
Élément de référence important, la règle de la variance est donnée par la relation u = C + 2 - j. On détermine les constantes d’équilibre par rapport à l’activité de chacun des constituants d’un mélange. Pourvu que les coefficients d’activité sont voisins de l’unité, les constantes d’équilibre peuvent s’exprimer en termes des concentrations, des fractions molaires. Lorsqu’un stress est imposé à un équilibre, la loi de LE CHATELIER - BRAUN permet de prévoir le sens de la modification de l’équilibre. L’ajout d’un gaz inerte est sans effet si la réaction a lieu à volume constant. Par contre, son effet à pression constante, dépend de la variation positive ou négative du nombre de moles observées au cours de la réaction. Dans le cas d’une réaction hétérogène, l’activité d’une phase pure étant égale à l’unité, on ne tient compte que des pressions partielles (gaz) et des activités en solutions condensées. Les constantes d’équilibre varient en fonction de la température. Par exemple, Kp est lié à l’enthalpie et KC à l'énergie interne.
|
Pour en savoir un peu plus
Un site intéressant à consulter : http://www.edu.gov.mb.ca/m12/frpub/ped/sn/chimie_40s/docs/reg_4.pdf .
VAN T'HOFF : http://nobelprizes.com/nobel/chemistry/1901a.html
12.1. Formation de l'iodure d'hydrogène
On observe l’équilibre de BODENSTEIN : I2 + H2
![]() 2 HI. À 763,8 et à 666,8 K on obtient les constantes
d’équilibre KP : elles sont respectivement 60,80 et 45,62. Calculez la
variation d’enthalpie de cette réaction (on négligera la variation de DH avec la température
puisque DT < 100 ºC).
2 HI. À 763,8 et à 666,8 K on obtient les constantes
d’équilibre KP : elles sont respectivement 60,80 et 45,62. Calculez la
variation d’enthalpie de cette réaction (on négligera la variation de DH avec la température
puisque DT < 100 ºC).
Réponse : DH = -12,5 kJ·mol-1.
12.2. Réduction de la vapeur d'eau par le carbone
L’étude des réactions chimiques entre le carbone et le gaz carbonique montre les résultats suivants :
[1] C (s) + CO2 (g) ![]() 2 CO (g) ; (équilibre de BOUDOUARD)
2 CO (g) ; (équilibre de BOUDOUARD)
R T log KP1 = - 40 900 + 4,9 T log T - 4,95 10-3 T2 + 3,1 10-7 T3 + 12,66 T
[2] CO2 (g) + H2 (g) ![]() CO (g)
+ H2O (g)
CO (g)
+ H2O (g)
R T log KP2 = - 10 100 + 1,81 T log T - 4,45 10-3 T2 + 4,8 10-7 T3 + 0,54 T
[3] C (s) + H2O (g) ![]() CO (g) + H2
(g).
CO (g) + H2
(g).
En utilisant le principe de l’état initial et de l’état final, déterminer la valeur de R T log KP3 en fonction de la température T.
Réponse : RT log KP3 = -30 800 + 3,09 T log T - 0,50 10-3 T2 - 1,7 10-7 T3 + 12,126 T
12.3. Dissociation de PCl5
Le pentachlorure de phosphores, PCl5, se dissocie selon la réaction :
PCl5 (g) ![]() PCl3 (g) + Cl2 (g)
PCl3 (g) + Cl2 (g)
a- À 250 °C, KP = 1,78 atm. Trouvez la fraction a de PCl5 qui est dissociée, si la réaction se déroule à la pression atmosphérique.
Réponse : a = 0,444
b- On recommence la même expérience mais cette fois dans un ballon fermé. Une mole de PCl5 y est introduite sous une pression initiale de 1 atm. Calculez la pression finale ainsi que la faction a' de PCl5 qui est dissocié.
Réponse : a' = 0,714
c- Cette décomposition est étudiée sous la pression atmosphérique et à 403 K. À cette température, le mélange obtenu à partir de PCl5 pur a une densité de 4,80 g/dm3. Calculez le degré de dissociation a de PCl5 et la valeur de DG à cette température. Recalculez la valeur de a si la pression totale est toujours de 1 atmosphère mais en présence d’une pression partielle d’argon de 0,5 atmosphères. Pouvez-vous réconcilier ces résultats et la loi de Lechatelier-Brown ?
12.4. Dissociation de l’anhydride sulfurique
Soit la réaction :
[2] 2 SO3 (g) ![]() 2 SO2 (g) + O2
(g)
2 SO2 (g) + O2
(g)
avec |
|
= 183,1 kJ mol-1 et |
|
= 132,9 kJ mol-1. |
Quel serait le rendement (% d’anhydride sulfureux transformé en SO3) de la réaction si on introduit un mélange comportant 8 % de SO2, 10% d’oxygène et 82 % d’azote dans un réacteur opérant à 450 K et sous la pression atmosphérique?
Réponse : La réaction est complète.
12.5. Oxydation de l’argent métallique
Le problème de l’oxydation de l’argent métallique est bien connu des propriétaires de coutellerie argentée : ils doivent périodiquement nettoyer les ustensiles qui noircissent en se recouvrant d’oxyde d’argent. Existe-t-il des conditions de température propres à la conservation de l’argenterie. Soit donc l’équilibre :
Ag2O (s) ![]() 2 Ag (s) + 1/2 O2 (g)
2 Ag (s) + 1/2 O2 (g)
On donne les valeurs suivantes :
Composé |
Ag2O (s) |
Ag (s) |
O2 (g) |
DH (kJ·mol-1) |
- 30,56 |
0 |
0 |
DSo (J·K-1·mol-1) |
121,60 |
42,68 |
204,82 |
1- Calculer la pression de l’oxygène à 298 K à l’équilibre
La capacité calorifique des constituants de l’équilibre est donnée par la relation :
|
= a + b (10-3) T + d (10+5) T-2 J·K-1·mol-1 |
Composé |
Ag2O (s) |
Ag (s) |
O2 (g) |
a |
55,43 |
21,27 |
34,57 |
b |
29,43 |
4,26 |
1,08 |
d |
- |
1,50 |
-7,86 |
Réponse : PO2 = 0,000 166 atm = 0,126 mmHg.
2- Calculer la valeur de DCP(réaction) en fonction de la température T.
Réponse : DCp(réaction) = 4,39 - 20,37 10-3 T - 0,93 105 T-2
| 3- Calculer la valeur de |
|
en fonction de la température T. |
4- En déduire les valeurs de DHo à 100, 200, 300, 400, 500, 600 et 700 K.
| 5- Calculer la valeur de |
|
en fonction de la température T. |
6- En déduire les valeurs de DSo à 100, 200, 300, 400, 500, 600 et 700 K.
7- En déduire les valeurs de DGo à 100, 200, 300, 400, 500, 600 et 700 K.
8- En déduire la constante d’équilibre KP à 100, 200, 300, 400, 500, 600 et 700 K.
Dire comment varie KP avec l’augmentation de température.
9- Déterminer graphiquement la température d’équilibre lorsque l’argenterie est exposée à l’air?
Réponse : l’équilibre sera atteint vers 410 K, soit encore vers 137 °C.
Quelle conclusion en tirez-vous pour préserver l’argenterie de l’oxydation?
12.6. Procédé HABER
Soit la synthèse de l’ammoniac :
1/2 N2 (g) + 3/2 H2 (g) ® NH3 (g)
| On donne les valeurs de |
|
(x) = a + b T 10-3 + c T-2 105 |
x |
a |
b |
d |
|
S |
H2 |
+ 27,28 |
+ 3,26 |
+ 0,50 |
- |
130,59 |
N2 |
+ 28,58 |
+ 3,76 |
- 0,50 |
- |
191,5 |
NH3 |
+ 29,75 |
+ 25,10 |
- 1,55 |
- 46,11 |
192,5 |
| 1- Calculer la valeur de |
|
(réaction) . |
Réponse :
DHo(réaction) = -46,11 kJ/mol.| 2- Calculer |
|
en fonction de la température T. |
3- En déduire les valeurs de DHo à 400, 450, 500 et 550 ºC.
Réponse : -54,104 kJ/mol à 550 °C.
| 4- Calculer la valeur de |
|
de la réaction. |
| 5- Calculer la valeur de |
|
en fonction de la température T. |
6- En déduire les valeurs de DSo à 400, 450, 500 et 550 ºC.
Réponse : -116,4 J/mol/K à 550 °C.
7- En déduire les valeurs de DGo à 400, 450, 500 et 550 ºC.
Réponse : + 41,49 kJ/mol à 550 °C.
8- En déduire la constante d’équilibre thermodynamique à 400, 450, 500 et 550 ºC sous une pression d'une (1) atmosphère. En déduire le sens de la variation de la constante KP avec la température T.
Réponse : Kp = 0,002 33 (atm-1) à 550 °C.
9- En supposant que les gaz ont un comportement idéal, établir la relation
|
en fonction de la pression P. |
10- Calculer les valeurs de DG450 sous des pressions de 10, 50, 100 et 300 atmosphères.
11- En déduire la constante thermodynamique à l’équilibre à 450 ºC et sous 10, 50, 100 et 300 atmosphères ainsi que les compositions des phases gazeuses à l’équilibre en supposant le mélange initial stœchiométrique. En déduire le sens de la variation de KP quand la pression croît.
12- Aux pressions mentionnées la constante Kg = 0,99, 0,94, 0,88 et 0,69 sous les pressions respectives de 10, 50, 100 et 300 atmosphères. En déduire ce que devient le mélange stœchiométrique à ces pressions.
Réponse : à la pression de 300 atm et à 450 °C, 92 % du mélange sont transformés en ammoniac.
Voir un texte mi historique, mi scientifique relatif au procédé HABER : G. Emptoz, L'Actualité chimique, le journal de la Société chimique de France, La nouvelle chimie de l'azote dans l'entre-deux-guerres, N° 381, pages 40-46, (janvier 2014).
12.7. Équilibres entre sels et leurs hydrates
À 25 °C, les tensions de vapeur du système phosphate monoacide de sodium et l'eau, Na2HPO4 – H2O sont les suivantes :
| Équilibres | Pression de vapeur (Pa) | ||
| Na2HPO4,12H2O | Na2HPO4,7H2O + 7 H2O | 2550 | |
| Na2HPO4,7H2O | Na2HPO4,2H2O + 5 H2O | 1934 | |
| Na2HPO4,2H2O | Na2HPO4 + 2 H2O | 1306 | |
a- Sachant qu’à la température considérée, le phosphate bisodique anhydre et ses hydrates sont solides, en vous servant de la règle des phases, expliquez pourquoi la pression doit être constante lorsqu’il y a équilibre entre deux hydrates.
b- Qu’arrive-t-il si on augmente graduellement la pression au-delà de 2550 Pa?
c- Quel hydrate sera stable à l’air et à 25 °C si la pression partielle de la vapeur d’eau est de 1400 Pa?
12.8. Fabrication du carbure de molybdène
Quelle pression de méthane faut-il ajouter au-dessus du molybdène à 973 K pour obtenir une pression partielle d’hydrogène de 76 kPa? On donne la valeur de la constante d’équilibre pour la réaction suivante :
|
2 Mo (sol) + CH4 (gaz) |
|
Mo2C (sol) + 2 H2 (gaz); |
Kp = 360 kPa |
Réponse : 53 822 Pa.
12.9. Déshydratation du cuivre trihydraté
Soit la réaction de déshydratation suivante :
|
CuSO4,3H2O |
|
CuSO4,H2O + 2 H2O(gaz) |
On observe que la pression de dissociation est de 746,8 Pa à 298,2 K et on sait par ailleurs que la variation de l’enthalpie sous la pression atmosphérique (101,333 kPa) et à 0 °C est de 112,86 kJ. On considérera cette valeur indépendante de la température.
Quelle sera la pression de dissociation à 100 °C ?
Quelle sera la température de dissociation sous une pression de 101333 Pa de vapeur d’eau ?
Réponse : T = 107 °C
Que pouvez-vous dire ou conclure de la température de dissociation dans l’air ambiant ?
12.10. Dissociation du dioxyde de carbone
À 3000 K et sous la pression atmosphérique (~101 333 Pa), 40 % du dioxyde de carbone est dissocié en monoxyde de carbone et en oxygène moléculaire. Quelle sera le degré de dissociation si la pression est élevée à 2 atmosphères ?
Réponse : le degré de dissociation du CO2 à 3000 K sous 2 atm est 33,5 %.
Quel sera le degré de dissociation de CO2 lorsqu’un mélange de 50 % de CO2 et de 50 % de O2 est porté à 3000 K sous la pression atmosphérique ?
12.11. Décomposition thermique du carbonate de calcium
Soit la réaction :
|
CaCO3 (sol) |
|
CaO (sol) + CO2 (gaz) |
On sait que pour cette réaction : DHo° = + 182,875 et DGo° = 134,763 kJ/mol.
- Calculez pour cette réaction : la valeur de DSo°.
Réponse : DSo° = 164,2 J/(mol·degré).
- En admettant que la valeur de l’enthalpie est indépendante de la température, calcules la valeur de DGo°.
- On place 10 g de carbonate de calcium solide dans un récipient préalablement évacué à 800 K. Quelle sera la pression finale dans le réacteur.
Réponse : Kp(800) = 4,33 10-4 Pa.
Dernière mise à jour : 2021-07-08.
x x x x x x x x x x x