
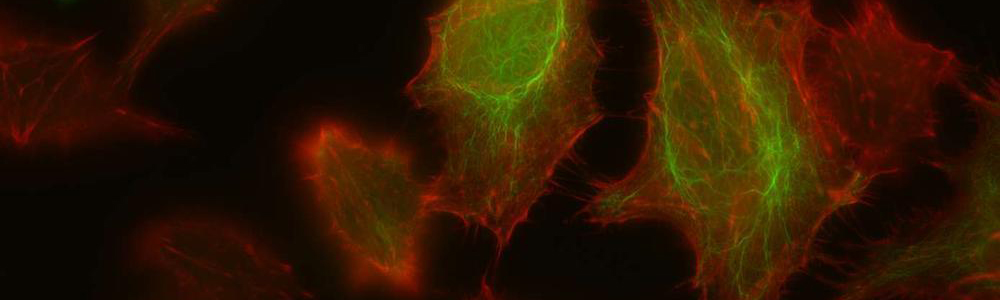


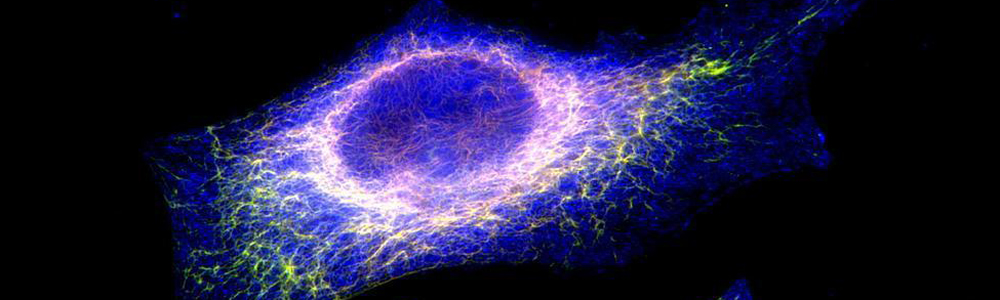


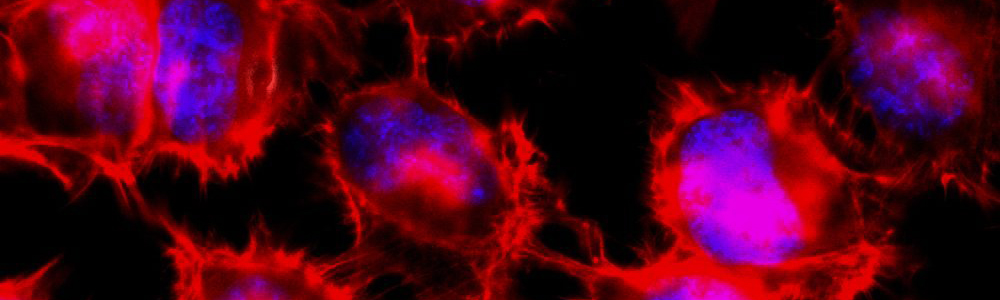
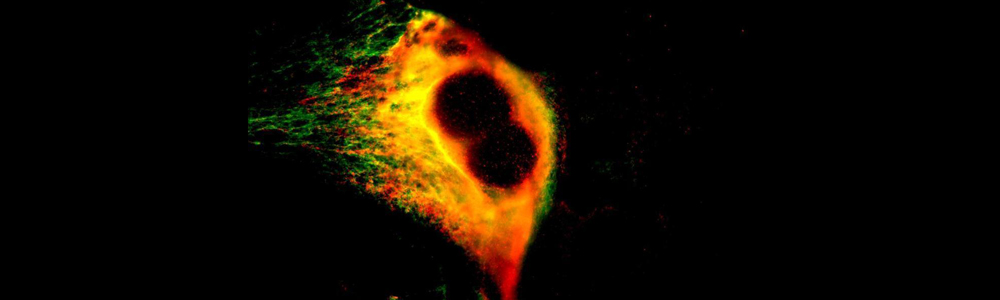
Epidermolyse bulleuse simplex (EBS)
Épidermolyse bulleuse simple (EBS)
L’EBS est une maladie génétique rare ayant une prévalence de 1 sur 50 000 à 1 sur 30 000. Cette maladie se caractérise par la formation de bulles ou d’ampoules cutanées suite à un frottement mécanique sur la peau. Ceci est occasionné par un décollement intra-épidermique au niveau de la couche basale de l’épiderme puisque certaines protéines contribuant à l’intégrité de la peau sont défectueuses. Cette maladie a un mode de transmission majoritairement autosomique dominant et est causée par plus de 100 mutations affectant les gènes des kératines 5 et 14 (KRT5 et KRT14) aboutissant à trois sous types : Weber–Cockayne, Koebner et Dowling-Meara (le plus sévère). Les mutations affectant les gènes KRT5 ou KRT14 conduisent à la synthèse de protéines défectueuses incapables de former un réseau de filaments organisés, ce qui entraînent la formation d’agrégats de kératine. Aucun traitement n’est disponible pour l’instant et seule une médication pour soulager la douleur et limiter les infections est disponible pour les personnes qui en sont atteintes.
Projets en cours :
- Étude de la phosphorylation du motif LLTPL K5 chez les individus atteints d’épidermolyse bulleuse simplex;
- Correction des mutations causales de l’épidermolyse bulleuse simplex grâce à la technologie CRISPR-Cas9 en vue d’obtenir des cellules saines pour les individus atteints et de procéder à des autogreffes de peau comme traitement curatif (Collaborateurs : Jacques P. Tremblay, Lucie Germain).
Site d’informations: http://www.debra-international.org/epidermolysis-bullosa.html
Agénésie Dysgénésie du corps calleux
Agénésie Dysgénésie du corps calleux
L’agénésie complète ou partielle du corps calleux peut être associée à un retard de développement, une hypotonie (tonus musculaire plus faible que la norme), une épilepsie et une microcéphalie. Ce phénotype a été observé par plusieurs pédiatres de la région du Saguenay-Lac-St-Jean (SLSJ). Ils ont noté que chez plusieurs enfants en bas âge présentant une agénésie ou une dysgénésie du corps calleux sans polyneuropathie associée avec une hypotonie, ceux-ci développaient par la suite un retard de développement, une épilepsie et une microcéphalie. Depuis cette observation clinique, les pédiatres du Centre intégré universitaire de santé et de services sociaux du SLSJ ont recensé 8 à 12 patients présentant cet aspect clinique. L’étude de ce phénotype précis par séquençage complet de l’exome des participants n’a pas permis d’identifier une mutation génétique commune. Toutefois, il a été possible de mettre en évidence des mutations au niveau de gènes impliqués dans certaines anomalies cérébrales présentées chez les participants, soit DCLK2, HERC2, KCNH3 et CACNA1A. Ces résultats représentent donc de nouvelles pistes à investiguer afin de mieux comprendre les causes génétiques derrière cette condition. La Pre Laprise dispose d’échantillons pour 4 de ces patients ainsi que pour leur famille (parents et frères) pour un total de 14 participants.
Acidose lactique
Acidose lactique
Le Syndrome de Leigh type Canadien-Français (SLCF) fait partie de la grande famille des syndromes de Leigh et porte également le nom d’acidose lactique congénitale de type Saguenay-Lac-St-Jean (SLSJ) qui se réfère à une crise acidotique qui peut survenir chez les enfants atteints. Dans la plupart des cas, cette crise aura lieu dans les cinq premières années de vie de l’enfant et sera mortelle. Dans la région du SLSJ, 1 personne sur 23 est porteuse et un enfant sur 2000 naissances est atteint. Les manifestations cliniques les plus fréquentes de cette maladie sont une hypotonie (tonus musculaire plus faible que la norme) et un retard du développement psychomoteur. Bien que ces manifestations soient observées chez la plupart des enfants atteints de SLCF, la variabilité du phénotype et du degré de sévérité est très importante d’un patient à l’autre. Certains enfants sont très actifs et marchent normalement alors que d’autres souffrent d’handicap physique plus lourd et se déplacent en fauteuil roulant. Le gène responsable de la maladie, leucine-rich pentatricopeptide repeat containing (LRPPRC) a été identifié en 2003. Il cause un déficit dans le fonctionnement du cytochrome c oxydase (COX) de la chaîne respiratoire des mitochondries. Les principaux organes touchés par cette maladie sont le foie et le cerveau pour lesquels un déficit de 90% de la protéine COX est observé. Il n’existe actuellement aucun traitement pour cette maladie mais certaines stratégies peuvent permettre une meilleure qualité de vie aux enfants atteints et prévenir les crises d’acidoses, comme des petits repas et une activité physique limitée.