




Epidermolysis bullosa simplex (EBS)
Epidermolysis bullosa simplex (EBS)
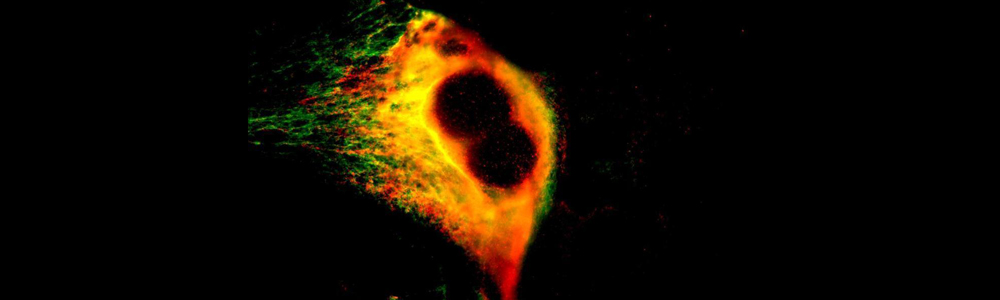
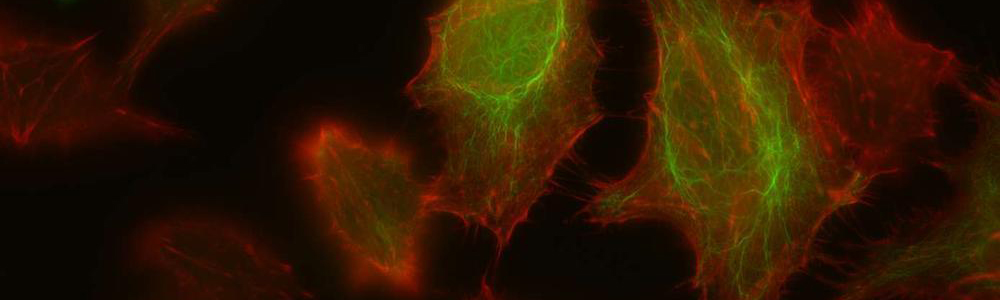
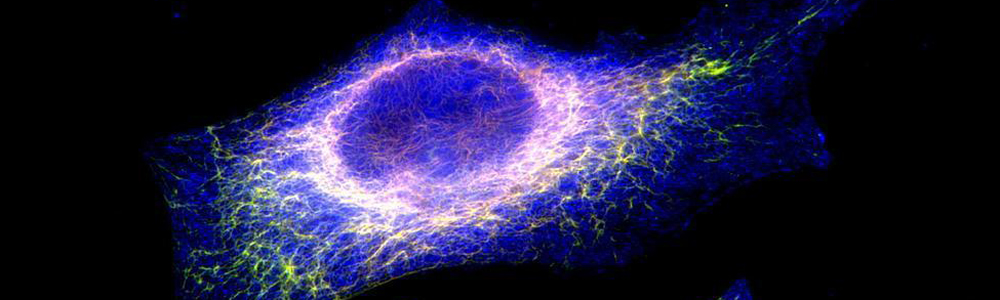
EBS is a rare genetic disease with a prevalence of 1 individual affected per 50,000 to 30,000 people. This disease is characterized by the formation of blisters on the skin following mechanical friction. This is caused by a lack on integrity in-between the basal layer of the epidermidis due to certain defective proteins of the skin. This disease has an autosomal dominant transmission mode and can be caused by more than a hundred mutations affecting keratin 5 and 14 genes (KRT5 and KRT14) resulting in three subtypes: Weber–Cockayne, Koebner et Dowling-Meara (the most severe form). The mutations in KRT4 and KRT15 lead to the synthesis of defective proteins which are unable to form an organized network of filaments, leading to the formation of keratin aggregates. No treatment is currently available and only medication to relieve pain and prevent infections is available for the affected individuals.
Ongoing research projects
- Investigation of the phophorylation of LLTPL K5 pattern in individuals with epidermolysis bullosa simplex;
- Correction of causal mutations in epidermolysis bullosa simplex with CRISPR-Cas9 technology in order to obtain healthy cells for affected individuals and to proceed to skin graft (Collaborators: Jacques P. Tremblay, Lucie Germain).
Information website: http://www.debra-international.org/epidermolysis-bullosa.html
Agenesis and digenesis of the corpus callosum
Agenesis and digenesis of the corpus callosum
Partial of complete agenesis of the corpus callosum can be associated with developmental delay, hypotonia (lower muscle tone than average), epilepsy and microcephaly. This phenotype was observed by several pediatrician of the Saguenay─Lac-Saint-Jean region (SLSJ). They noted that several young children with an agenesis or disgenesia of the corpus callosum without polyneuropathy associated to hypotonia, were also developing a developmental delay, epilepsy and microcephaly. Since this initial observation, pediatricians of the Centre intégré universitaire de santé et de services sociaux du SLSJ identified 8 to 12 patients with this particular clinical profile. The study of this particular phenotype was assessed by complete exome sequencing of affected individuals but did not identified a common genetic defect. However, several mutations in genes involved in cerebral anomalies were identified in participants which are DCLK2, HERC2, KCNH3 et CACNA1A. These results represent new lead of investigation to follow in order to understand better the genetic causes behind this condition. Prof Laprise dispose of samples for 4 of these patients and their families (parents and brothers) for a total of 14 research participants.
French Canadian Leigh Syndrome
French Canadian Leigh Syndrome
Leigh syndrome French Canadian (LSFC) is a neurodegenerative disorder which is part of the large family of Leigh syndromes and may also be called congenital lactic acidosis type Saguenay─Lac-St-Jean (SLSJ) which refers to the acidotic crisis that may happen during childhood. In most cases, this crisis will be in the 5 first year of life and will be fatal. In the SLSJ region, the carrier rate is 1/23 and 1 child over 2000 births is affected. The most frequent clinical manifestations are hypotonia (lower muscle tone than average) and a delayed neuromotor development. Although these events are observed in most children with LFSC, the variability of the phenotype and the severity is very important among the patients. Some children are very active and walk normally while others have severe physical disabilities and need to use a wheelchair. LSFC is caused by a mutation in the Leucin-rich pentatricopeptide repeat containing (LRPPRC) gene. This mutation leads to a deficit in the cytochrome c oxydase (COX) deficiency in the complex IV of the mitochondria respiratory chain. The main organs affected by this deficiency are the brain and liver in which a 90% deficit for the COX protein is present. At the moment, no cure exists for this disease but care for an improved quality of life are used in order to prevent acidotic crisis, such as small meals and limited physical activities.